Documentation for COBRApy¶
For installation instructions, please see INSTALL.rst.
Many of the examples below are viewable as IPython notebooks, which can be viewed at nbviewer.
Getting Started¶
To begin with, cobrapy comes with bundled models for Salmonella and E. coli, as well as a “textbook” model of E. coli core metabolism. To load a test model, type
In [1]:
from __future__ import print_function
import cobra.test
# "ecoli" and "salmonella" are also valid arguments
model = cobra.test.create_test_model("textbook")
The reactions, metabolites, and genes attributes of the cobrapy model are a special type of list called a DictList, and each one is made up of Reaction, Metabolite and Gene objects respectively.
In [2]:
print(len(model.reactions))
print(len(model.metabolites))
print(len(model.genes))
95
72
137
Just like a regular list, objects in the DictList can be retrived by index. For example, to get the 30th reaction in the model (at index 29 because of 0-indexing):
In [3]:
model.reactions[29]
Out[3]:
<Reaction EX_glu__L_e at 0x7f56b0ea3198>
Addictionally, items can be retrived by their id using the get_by_id() function. For example, to get the cytosolic atp metabolite object (the id is “atp_c”), we can do the following:
In [4]:
model.metabolites.get_by_id("atp_c")
Out[4]:
<Metabolite atp_c at 0x7f56b0ed7cc0>
As an added bonus, users with an interactive shell such as IPython will be able to tab-complete to list elements inside a list. While this is not recommended behavior for most code because of the possibility for characters like “-” inside ids, this is very useful while in an interactive prompt:
In [5]:
model.reactions.EX_glc__D_e.lower_bound
Out[5]:
-10.0
Reactions¶
We will consider the reaction glucose 6-phosphate isomerase, which interconverts glucose 6-phosphate and fructose 6-phosphate. The reaction id for this reaction in our test model is PGI.
In [6]:
pgi = model.reactions.get_by_id("PGI")
pgi
Out[6]:
<Reaction PGI at 0x7f56b0e396d8>
We can view the full name and reaction catalyzed as strings
In [7]:
print(pgi.name)
print(pgi.reaction)
glucose-6-phosphate isomerase
g6p_c <=> f6p_c
We can also view reaction upper and lower bounds. Because the pgi.lower_bound < 0, and pgi.upper_bound > 0, pgi is reversible
In [8]:
print(pgi.lower_bound, "< pgi <", pgi.upper_bound)
print(pgi.reversibility)
-1000.0 < pgi < 1000.0
True
We can also ensure the reaction is mass balanced. This function will return elements which violate mass balance. If it comes back empty, then the reaction is mass balanced.
In [9]:
pgi.check_mass_balance()
Out[9]:
{}
In order to add a metabolite, we pass in a dict with the metabolite object and its coefficient
In [10]:
pgi.add_metabolites({model.metabolites.get_by_id("h_c"): -1})
pgi.reaction
Out[10]:
'g6p_c + h_c <=> f6p_c'
The reaction is no longer mass balanced
In [11]:
pgi.check_mass_balance()
Out[11]:
{'H': -1.0, 'charge': -1.0}
We can remove the metabolite, and the reaction will be balanced once again.
In [12]:
pgi.pop(model.metabolites.get_by_id("h_c"))
print(pgi.reaction)
print(pgi.check_mass_balance())
g6p_c <=> f6p_c
{}
It is also possible to build the reaction from a string. However, care must be taken when doing this to ensure reaction id’s match those in the model. The direction of the arrow is also used to update the upper and lower bounds.
In [13]:
pgi.reaction = "g6p_c --> f6p_c + h_c + green_eggs + ham"
unknown metabolite 'green_eggs' created
unknown metabolite 'ham' created
In [14]:
pgi.reaction
Out[14]:
'g6p_c --> f6p_c + green_eggs + h_c + ham'
In [15]:
pgi.reaction = "g6p_c <=> f6p_c"
pgi.reaction
Out[15]:
'g6p_c <=> f6p_c'
Metabolites¶
We will consider cytosolic atp as our metabolite, which has the id atp_c in our test model.
In [16]:
atp = model.metabolites.get_by_id("atp_c")
atp
Out[16]:
<Metabolite atp_c at 0x7f56b0ed7cc0>
We can print out the metabolite name and compartment (cytosol in this case).
In [17]:
print(atp.name)
print(atp.compartment)
ATP
c
We can see that ATP is a charged molecule in our model.
In [18]:
atp.charge
Out[18]:
-4
We can see the chemical formula for the metabolite as well.
In [19]:
print(atp.formula)
C10H12N5O13P3
The reactions attribute gives a frozenset of all reactions using the given metabolite. We can use this to count the number of reactions which use atp.
In [20]:
len(atp.reactions)
Out[20]:
13
A metabolite like glucose 6-phosphate will participate in fewer reactions.
In [21]:
model.metabolites.get_by_id("g6p_c").reactions
Out[21]:
frozenset({<Reaction GLCpts at 0x7f56b0eabcc0>,
<Reaction Biomass_Ecoli_core at 0x7f56b0e9e358>,
<Reaction G6PDH2r at 0x7f56b0eab9e8>,
<Reaction PGI at 0x7f56b0e396d8>})
Genes¶
The gene_reaction_rule is a boolean representation of the gene requirements for this reaction to be active as described in Schellenberger et al 2011 Nature Protocols 6(9):1290-307.
The GPR is stored as the gene_reaction_rule for a Reaction object as a string.
In [22]:
gpr = pgi.gene_reaction_rule
gpr
Out[22]:
'b4025'
Corresponding gene objects also exist. These objects are tracked by the reactions itself, as well as by the model
In [23]:
pgi.genes
Out[23]:
frozenset({<Gene b4025 at 0x7f56b0e8fac8>})
In [24]:
pgi_gene = model.genes.get_by_id("b4025")
pgi_gene
Out[24]:
<Gene b4025 at 0x7f56b0e8fac8>
Each gene keeps track of the reactions it catalyzes
In [25]:
pgi_gene.reactions
Out[25]:
frozenset({<Reaction PGI at 0x7f56b0e396d8>})
Altering the gene_reaction_rule will create new gene objects if necessary and update all relationships.
In [26]:
pgi.gene_reaction_rule = "(spam or eggs)"
pgi.genes
Out[26]:
frozenset({<Gene eggs at 0x7f56b0e35ba8>, <Gene spam at 0x7f56b0e390f0>})
In [27]:
pgi_gene.reactions
Out[27]:
frozenset()
Newly created genes are also added to the model
In [28]:
model.genes.get_by_id("spam")
Out[28]:
<Gene spam at 0x7f56b0e390f0>
The delete_model_genes function will evaluate the gpr and set the upper and lower bounds to 0 if the reaction is knocked out. This function can preserve existing deletions or reset them using the cumulative_deletions flag.
In [29]:
cobra.manipulation.delete_model_genes(model, ["spam"],
cumulative_deletions=True)
print("after 1 KO: %4d < flux_PGI < %4d" %
(pgi.lower_bound, pgi.upper_bound))
cobra.manipulation.delete_model_genes(model, ["eggs"],
cumulative_deletions=True)
print("after 2 KO: %4d < flux_PGI < %4d" %
(pgi.lower_bound, pgi.upper_bound))
after 1 KO: -1000 < flux_PGI < 1000
after 2 KO: 0 < flux_PGI < 0
The undelete_model_genes can be used to reset a gene deletion
In [30]:
cobra.manipulation.undelete_model_genes(model)
print(pgi.lower_bound, "< pgi <", pgi.upper_bound)
-1000 < pgi < 1000
Building a Model¶
This simple example demonstrates how to create a model, create a reaction, and then add the reaction to the model.
We’ll use the ‘3OAS140’ reaction from the STM_1.0 model:
1.0 malACP[c] + 1.0 h[c] + 1.0 ddcaACP[c] \(\rightarrow\) 1.0 co2[c] + 1.0 ACP[c] + 1.0 3omrsACP[c]
First, create the model and reaction.
In [1]:
from cobra import Model, Reaction, Metabolite
# Best practise: SBML compliant IDs
cobra_model = Model('example_cobra_model')
reaction = Reaction('3OAS140')
reaction.name = '3 oxoacyl acyl carrier protein synthase n C140 '
reaction.subsystem = 'Cell Envelope Biosynthesis'
reaction.lower_bound = 0. # This is the default
reaction.upper_bound = 1000. # This is the default
reaction.objective_coefficient = 0. # this is the default
We need to create metabolites as well. If we were using an existing model, we could use get_by_id to get the apporpriate Metabolite objects instead.
In [2]:
ACP_c = Metabolite(
'ACP_c',
formula='C11H21N2O7PRS',
name='acyl-carrier-protein',
compartment='c')
omrsACP_c = Metabolite(
'3omrsACP_c',
formula='C25H45N2O9PRS',
name='3-Oxotetradecanoyl-acyl-carrier-protein',
compartment='c')
co2_c = Metabolite(
'co2_c',
formula='CO2',
name='CO2',
compartment='c')
malACP_c = Metabolite(
'malACP_c',
formula='C14H22N2O10PRS',
name='Malonyl-acyl-carrier-protein',
compartment='c')
h_c = Metabolite(
'h_c',
formula='H',
name='H',
compartment='c')
ddcaACP_c = Metabolite(
'ddcaACP_c',
formula='C23H43N2O8PRS',
name='Dodecanoyl-ACP-n-C120ACP',
compartment='c')
Adding metabolites to a reaction requires using a dictionary of the metabolites and their stoichiometric coefficients. A group of metabolites can be added all at once, or they can be added one at a time.
In [3]:
reaction.add_metabolites({malACP_c: -1.0,
h_c: -1.0,
ddcaACP_c: -1.0,
co2_c: 1.0,
ACP_c: 1.0,
omrsACP_c: 1.0})
reaction.reaction # This gives a string representation of the reaction
Out[3]:
'ddcaACP_c + h_c + malACP_c --> 3omrsACP_c + ACP_c + co2_c'
The gene_reaction_rule is a boolean representation of the gene requirements for this reaction to be active as described in Schellenberger et al 2011 Nature Protocols 6(9):1290-307. We will assign the gene reaction rule string, which will automatically create the corresponding gene objects.
In [4]:
reaction.gene_reaction_rule = '( STM2378 or STM1197 )'
reaction.genes
Out[4]:
frozenset({<Gene STM2378 at 0x7fada4592908>, <Gene STM1197 at 0x7fada45927f0>})
At this point in time, the model is still empty
In [5]:
print('%i reactions initially' % len(cobra_model.reactions))
print('%i metabolites initially' % len(cobra_model.metabolites))
print('%i genes initially' % len(cobra_model.genes))
0 reactions initially
0 metabolites initially
0 genes initially
We will add the reaction to the model, which will also add all associated metabolites and genes
In [6]:
cobra_model.add_reaction(reaction)
# Now there are things in the model
print('%i reaction' % len(cobra_model.reactions))
print('%i metabolites' % len(cobra_model.metabolites))
print('%i genes' % len(cobra_model.genes))
1 reaction
6 metabolites
2 genes
We can iterate through the model objects to observe the contents
In [7]:
# Iterate through the the objects in the model
print("Reactions")
print("---------")
for x in cobra_model.reactions:
print("%s : %s" % (x.id, x.reaction))
print("")
print("Metabolites")
print("-----------")
for x in cobra_model.metabolites:
print('%9s : %s' % (x.id, x.formula))
print("")
print("Genes")
print("-----")
for x in cobra_model.genes:
associated_ids = (i.id for i in x.reactions)
print("%s is associated with reactions: %s" %
(x.id, "{" + ", ".join(associated_ids) + "}"))
Reactions
---------
3OAS140 : ddcaACP_c + h_c + malACP_c --> 3omrsACP_c + ACP_c + co2_c
Metabolites
-----------
3omrsACP_c : C25H45N2O9PRS
ddcaACP_c : C23H43N2O8PRS
ACP_c : C11H21N2O7PRS
co2_c : CO2
malACP_c : C14H22N2O10PRS
h_c : H
Genes
-----
STM2378 is associated with reactions: {3OAS140}
STM1197 is associated with reactions: {3OAS140}
Reading and Writing Models¶
Cobrapy supports reading and writing models in SBML (with and without FBC), JSON, MAT, and pickle formats. Generally, SBML with FBC version 2 is the preferred format for general use. The JSON format may be more useful for cobrapy-specific functionality.
The package also ships with test models in various formats for testing purposes.
In [1]:
import cobra.test
import os
from os.path import join
data_dir = cobra.test.data_directory
print("mini test files: ")
print(", ".join(i for i in os.listdir(data_dir)
if i.startswith("mini")))
textbook_model = cobra.test.create_test_model("textbook")
ecoli_model = cobra.test.create_test_model("ecoli")
salmonella_model = cobra.test.create_test_model("salmonella")
mini test files:
mini.mat, mini_cobra.xml, mini.json, mini_fbc2.xml.gz, mini_fbc2.xml.bz2, mini_fbc2.xml, mini_fbc1.xml, mini.pickle
SBML¶
The Systems Biology Markup Language is an XML-based standard format for distributing models which has support for COBRA models through the FBC extension version 2.
Cobrapy has native support for reading and writing SBML with FBCv2. Please note that all id’s in the model must conform to the SBML SID requirements in order to generate a valid SBML file.
In [2]:
cobra.io.read_sbml_model(join(data_dir, "mini_fbc2.xml"))
Out[2]:
<Model mini_textbook at 0x7fa5e44d1a58>
In [3]:
cobra.io.write_sbml_model(textbook_model, "test_fbc2.xml")
There are other dialects of SBML prior to FBC 2 which have previously been use to encode COBRA models. The primary ones is the “COBRA” dialect which used the “notes” fields in SBML files.
Cobrapy can use libsbml, which must be installed separately (see installation instructions) to read and write these files. When reading in a model, it will automatically detect whether fbc was used or not. When writing a model, the use_fbc_package flag can be used can be used to write files in this legacy “cobra” format.
In [4]:
cobra.io.read_sbml_model(join(data_dir, "mini_cobra.xml"))
Out[4]:
<Model mini_textbook at 0x7fa5ba4d12e8>
In [5]:
cobra.io.write_sbml_model(textbook_model, "test_cobra.xml",
use_fbc_package=False)
JSON¶
cobrapy models have a JSON (JavaScript Object Notation) representation. This format was crated for interoperability with escher.
In [6]:
cobra.io.load_json_model(join(data_dir, "mini.json"))
Out[6]:
<Model mini_textbook at 0x7fa5ba4a3128>
In [7]:
cobra.io.save_json_model(textbook_model, "test.json")
MATLAB¶
Often, models may be imported and exported soley for the purposes of working with the same models in cobrapy and the MATLAB cobra toolbox. MATLAB has its own ”.mat” format for storing variables. Reading and writing to these mat files from python requires scipy.
A mat file can contain multiple MATLAB variables. Therefore, the variable name of the model in the MATLAB file can be passed into the reading function:
In [8]:
cobra.io.load_matlab_model(join(data_dir, "mini.mat"),
variable_name="mini_textbook")
Out[8]:
<Model mini_textbook at 0x7fa5ba483198>
If the mat file contains only a single model, cobra can figure out which variable to read from, and the variable_name paramter is unnecessary.
In [9]:
cobra.io.load_matlab_model(join(data_dir, "mini.mat"))
Out[9]:
<Model mini_textbook at 0x7fa5ba4a3f28>
Saving models to mat files is also relatively straightforward
In [10]:
cobra.io.save_matlab_model(textbook_model, "test.mat")
Simulating with FBA¶
Simulations using flux balance analysis can be solved using Model.optimize(). This will maximize or minimize (maximizing is the default) flux through the objective reactions.
In [1]:
import pandas
pandas.options.display.max_rows = 100
import cobra.test
model = cobra.test.create_test_model("textbook")
Running FBA¶
In [2]:
model.optimize()
Out[2]:
<Solution 0.87 at 0x10ddd0080>
The Model.optimize() function will return a Solution object, which will also be stored at model.solution. A solution object has several attributes:
- f: the objective value
- status: the status from the linear programming solver
- x_dict: a dictionary of {reaction_id: flux_value} (also called “primal”)
- x: a list for x_dict
- y_dict: a dictionary of {metabolite_id: dual_value}.
- y: a list for y_dict
For example, after the last call to model.optimize(), the status should be ‘optimal’ if the solver returned no errors, and f should be the objective value
In [3]:
model.solution.status
Out[3]:
'optimal'
In [4]:
model.solution.f
Out[4]:
0.8739215069684305
Analyzing FBA solutions¶
Models solved using FBA can be further analyzed by using summary methods, which output printed text to give a quick representation of model behavior. Calling the summary method on the entire model displays information on the input and output behavior of the model, along with the optimized objective.
In [5]:
model.summary()
IN FLUXES OUT FLUXES OBJECTIVES
o2_e -21.80 h2o_e 29.18 Biomass_Ecoli_core 0.874
glc__D_e -10.00 co2_e 22.81
nh4_e -4.77 h_e 17.53
pi_e -3.21
In addition, the input-output behavior of individual metabolites can also be inspected using summary methods. For instance, the following commands can be used to examine the overall redox balance of the model
In [6]:
model.metabolites.nadh_c.summary()
PRODUCING REACTIONS -- Nicotinamide adenine dinucleotide - reduced
------------------------------------------------------------------
% FLUX RXN ID REACTION
41.6% 16 GAPD g3p_c + nad_c + pi_c <=> 13dpg_c + h_c + nadh_c
24.1% 9.3 PDH coa_c + nad_c + pyr_c --> accoa_c + co2_c + nadh_c
13.1% 5.1 AKGDH akg_c + coa_c + nad_c --> co2_c + nadh_c + succoa_c
13.1% 5.1 MDH mal__L_c + nad_c <=> h_c + nadh_c + oaa_c
8.0% 3.1 Bioma... 1.496 3pg_c + 3.7478 accoa_c + 59.81 atp_c + 0.36...
CONSUMING REACTIONS -- Nicotinamide adenine dinucleotide - reduced
------------------------------------------------------------------
% FLUX RXN ID REACTION
100.0% -39 NADH16 4.0 h_c + nadh_c + q8_c --> 3.0 h_e + nad_c + q8h2_c
Or to get a sense of the main energy production and consumption reactions
In [7]:
model.metabolites.atp_c.summary()
PRODUCING REACTIONS -- ATP
--------------------------
% FLUX RXN ID REACTION
66.6% 46 ATPS4r adp_c + 4.0 h_e + pi_c <=> atp_c + h2o_c + 3.0 h_c
23.4% 16 PGK 3pg_c + atp_c <=> 13dpg_c + adp_c
7.4% 5.1 SUCOAS atp_c + coa_c + succ_c <=> adp_c + pi_c + succoa_c
2.6% 1.8 PYK adp_c + h_c + pep_c --> atp_c + pyr_c
CONSUMING REACTIONS -- ATP
--------------------------
% FLUX RXN ID REACTION
76.5% -52 Bioma... 1.496 3pg_c + 3.7478 accoa_c + 59.81 atp_c + 0.36...
12.3% -8.4 ATPM atp_c + h2o_c --> adp_c + h_c + pi_c
10.9% -7.5 PFK atp_c + f6p_c --> adp_c + fdp_c + h_c
Changing the Objectives¶
The objective function is determined from the objective_coefficient attribute of the objective reaction(s). Generally, a “biomass” function which describes the composition of metabolites which make up a cell is used.
In [8]:
biomass_rxn = model.reactions.get_by_id("Biomass_Ecoli_core")
Currently in the model, there is only one objective reaction (the biomass reaction), with an objective coefficient of 1.
In [9]:
model.objective
Out[9]:
{<Reaction Biomass_Ecoli_core at 0x116510828>: 1.0}
The objective function can be changed by assigning Model.objective, which can be a reaction object (or just it’s name), or a dict of {Reaction: objective_coefficient}.
In [10]:
# change the objective to ATPM
model.objective = "ATPM"
# The upper bound should be 1000, so that we get
# the actual optimal value
model.reactions.get_by_id("ATPM").upper_bound = 1000.
model.objective
Out[10]:
{<Reaction ATPM at 0x1165107b8>: 1}
In [11]:
model.optimize().f
Out[11]:
174.99999999999997
The objective function can also be changed by setting Reaction.objective_coefficient directly.
In [12]:
model.reactions.get_by_id("ATPM").objective_coefficient = 0.
biomass_rxn.objective_coefficient = 1.
model.objective
Out[12]:
{<Reaction Biomass_Ecoli_core at 0x116510828>: 1.0}
Running FVA¶
FBA will not give always give unique solution, because multiple flux states can achieve the same optimum. FVA (or flux variability analysis) finds the ranges of each metabolic flux at the optimum.
In [13]:
fva_result = cobra.flux_analysis.flux_variability_analysis(
model, model.reactions[:20])
pandas.DataFrame.from_dict(fva_result).T.round(5)
Out[13]:
| maximum | minimum | |
|---|---|---|
| ACALD | 0.00000 | 0.00000 |
| ACALDt | -0.00000 | 0.00000 |
| ACKr | -0.00000 | 0.00000 |
| ACONTa | 6.00725 | 6.00725 |
| ACONTb | 6.00725 | 6.00725 |
| ACt2r | 0.00000 | 0.00000 |
| ADK1 | -0.00000 | 0.00000 |
| AKGDH | 5.06438 | 5.06438 |
| AKGt2r | 0.00000 | 0.00000 |
| ALCD2x | 0.00000 | 0.00000 |
| ATPM | 8.39000 | 8.39000 |
| ATPS4r | 45.51401 | 45.51401 |
| Biomass_Ecoli_core | 0.87392 | 0.87392 |
| CO2t | -22.80983 | -22.80983 |
| CS | 6.00725 | 6.00725 |
| CYTBD | 43.59899 | 43.59899 |
| D_LACt2 | 0.00000 | 0.00000 |
| ENO | 14.71614 | 14.71614 |
| ETOHt2r | 0.00000 | 0.00000 |
| EX_ac_e | -0.00000 | 0.00000 |
Setting parameter fraction_of_optimium=0.90 would give the flux ranges for reactions at 90% optimality.
In [14]:
fva_result = cobra.flux_analysis.flux_variability_analysis(
model, model.reactions[:20], fraction_of_optimum=0.9)
pandas.DataFrame.from_dict(fva_result).T.round(5)
Out[14]:
| maximum | minimum | |
|---|---|---|
| ACALD | 0.00000 | -2.54237 |
| ACALDt | -0.00000 | -2.54237 |
| ACKr | -0.00000 | -3.81356 |
| ACONTa | 8.89452 | 0.84859 |
| ACONTb | 8.89452 | 0.84859 |
| ACt2r | 0.00000 | -3.81356 |
| ADK1 | 17.16100 | 0.00000 |
| AKGDH | 8.04593 | 0.00000 |
| AKGt2r | 0.00000 | -1.43008 |
| ALCD2x | 0.00000 | -2.21432 |
| ATPM | 25.55100 | 8.39000 |
| ATPS4r | 59.38106 | 34.82562 |
| Biomass_Ecoli_core | 0.87392 | 0.78653 |
| CO2t | -15.20653 | -26.52885 |
| CS | 8.89452 | 0.84859 |
| CYTBD | 51.23909 | 35.98486 |
| D_LACt2 | 0.00000 | -2.14512 |
| ENO | 16.73252 | 8.68659 |
| ETOHt2r | 0.00000 | -2.21432 |
| EX_ac_e | 3.81356 | 0.00000 |
Running FVA in summary methods¶
Flux variability analysis can also be embedded in calls to summary methods. For instance, the expected variability in substrate consumption and product formation can be quickly found by
In [15]:
model.optimize()
model.summary(fva=0.95)
IN FLUXES OUT FLUXES OBJECTIVES
o2_e -21.80 ± 1.91 h2o_e 27.86 ± 2.86 Biomass_Ecoli_core 0.874
glc__D_e -9.76 ± 0.24 co2_e 21.81 ± 2.86
nh4_e -4.84 ± 0.32 h_e 19.51 ± 2.86
pi_e -3.13 ± 0.08 for_e 2.86 ± 2.86
ac_e 0.95 ± 0.95
acald_e 0.64 ± 0.64
pyr_e 0.64 ± 0.64
etoh_e 0.55 ± 0.55
lac__D_e 0.54 ± 0.54
succ_e 0.42 ± 0.42
akg_e 0.36 ± 0.36
glu__L_e 0.32 ± 0.32
Similarly, variability in metabolite mass balances can also be checked with flux variability analysis
In [16]:
model.metabolites.pyr_c.summary(fva=0.95)
PRODUCING REACTIONS -- Pyruvate
-------------------------------
% FLUX RXN ID REACTION
85.0% 9.76 ± 0.24 GLCpts glc__D_e + pep_c --> g6p_c + pyr_c
15.0% 6.13 ± 6.13 PYK adp_c + h_c + pep_c --> atp_c + pyr_c
CONSUMING REACTIONS -- Pyruvate
-------------------------------
% FLUX RXN ID REACTION
78.9% 11.34 ± 7.43 PDH coa_c + nad_c + pyr_c --> accoa_c + co2_c + nadh_c
21.1% 0.85 ± 0.02 Bioma... 1.496 3pg_c + 3.7478 accoa_c + 59.81 atp_c + 0.36...
In these summary methods, the values are reported as a the center point +/- the range of the FVA solution, calculated from the maximum and minimum values.
Running pFBA¶
Parsimonious FBA (often written pFBA) finds a flux distribution which gives the optimal growth rate, but minimizes the total sum of flux. This involves solving two sequential linear programs, but is handled transparently by cobrapy. For more details on pFBA, please see Lewis et al. (2010).
In [17]:
FBA_sol = model.optimize()
pFBA_sol = cobra.flux_analysis.optimize_minimal_flux(model)
These functions should give approximately the same objective value
In [18]:
abs(FBA_sol.f - pFBA_sol.f)
Out[18]:
1.1102230246251565e-16
Simulating Deletions¶
In [1]:
import pandas
from time import time
import cobra.test
from cobra.flux_analysis import \
single_gene_deletion, single_reaction_deletion, \
double_gene_deletion, double_reaction_deletion
cobra_model = cobra.test.create_test_model("textbook")
ecoli_model = cobra.test.create_test_model("ecoli")
Single Deletions¶
Perform all single gene deletions on a model
In [2]:
growth_rates, statuses = single_gene_deletion(cobra_model)
These can also be done for only a subset of genes
In [3]:
gr, st = single_gene_deletion(cobra_model,
cobra_model.genes[:20])
pandas.DataFrame.from_dict({"growth_rates": gr,
"status": st})
Out[3]:
| growth_rates | status | |
|---|---|---|
| b0116 | 0.782351 | optimal |
| b0118 | 0.873922 | optimal |
| b0351 | 0.873922 | optimal |
| b0356 | 0.873922 | optimal |
| b0474 | 0.873922 | optimal |
| b0726 | 0.858307 | optimal |
| b0727 | 0.858307 | optimal |
| b1241 | 0.873922 | optimal |
| b1276 | 0.873922 | optimal |
| b1478 | 0.873922 | optimal |
| b1849 | 0.873922 | optimal |
| b2296 | 0.873922 | optimal |
| b2587 | 0.873922 | optimal |
| b3115 | 0.873922 | optimal |
| b3732 | 0.374230 | optimal |
| b3733 | 0.374230 | optimal |
| b3734 | 0.374230 | optimal |
| b3735 | 0.374230 | optimal |
| b3736 | 0.374230 | optimal |
| s0001 | 0.211141 | optimal |
This can also be done for reactions
In [4]:
gr, st = single_reaction_deletion(cobra_model,
cobra_model.reactions[:20])
pandas.DataFrame.from_dict({"growth_rates": gr,
"status": st}).round(4)
Out[4]:
| growth_rates | status | |
|---|---|---|
| ACALD | 0.8739 | optimal |
| ACALDt | 0.8739 | optimal |
| ACKr | 0.8739 | optimal |
| ACONTa | 0.0000 | optimal |
| ACONTb | 0.0000 | optimal |
| ACt2r | 0.8739 | optimal |
| ADK1 | 0.8739 | optimal |
| AKGDH | 0.8583 | optimal |
| AKGt2r | 0.8739 | optimal |
| ALCD2x | 0.8739 | optimal |
| ATPM | 0.9166 | optimal |
| ATPS4r | 0.3742 | optimal |
| Biomass_Ecoli_core | 0.0000 | optimal |
| CO2t | 0.4617 | optimal |
| CS | -0.0000 | optimal |
| CYTBD | 0.2117 | optimal |
| D_LACt2 | 0.8739 | optimal |
| ENO | -0.0000 | optimal |
| ETOHt2r | 0.8739 | optimal |
| EX_ac_e | 0.8739 | optimal |
Double Deletions¶
Double deletions run in a similar way. Passing in return_frame=True will cause them to format the results as a pandas Dataframe
In [5]:
double_gene_deletion(cobra_model, cobra_model.genes[-5:],
return_frame=True).round(4)
Out[5]:
| b2464 | b0008 | b2935 | b2465 | b3919 | |
|---|---|---|---|---|---|
| b2464 | 0.8739 | 0.8648 | 0.8739 | 0.8739 | 0.704 |
| b0008 | 0.8648 | 0.8739 | 0.8739 | 0.8739 | 0.704 |
| b2935 | 0.8739 | 0.8739 | 0.8739 | 0.0000 | 0.704 |
| b2465 | 0.8739 | 0.8739 | 0.0000 | 0.8739 | 0.704 |
| b3919 | 0.7040 | 0.7040 | 0.7040 | 0.7040 | 0.704 |
By default, the double deletion function will automatically use multiprocessing, splitting the task over up to 4 cores if they are available. The number of cores can be manually sepcified as well. Setting use of a single core will disable use of the multiprocessing library, which often aids debuggging.
In [6]:
start = time() # start timer()
double_gene_deletion(ecoli_model, ecoli_model.genes[:300],
number_of_processes=2)
t1 = time() - start
print("Double gene deletions for 200 genes completed in "
"%.2f sec with 2 cores" % t1)
start = time() # start timer()
double_gene_deletion(ecoli_model, ecoli_model.genes[:300],
number_of_processes=1)
t2 = time() - start
print("Double gene deletions for 200 genes completed in "
"%.2f sec with 1 core" % t2)
print("Speedup of %.2fx" % (t2 / t1))
Double gene deletions for 200 genes completed in 27.03 sec with 2 cores
Double gene deletions for 200 genes completed in 40.73 sec with 1 core
Speedup of 1.51x
Double deletions can also be run for reactions
In [7]:
double_reaction_deletion(cobra_model,
cobra_model.reactions[2:7],
return_frame=True).round(4)
Out[7]:
| ACKr | ACONTa | ACONTb | ACt2r | ADK1 | |
|---|---|---|---|---|---|
| ACKr | 0.8739 | 0.0 | 0.0 | 0.8739 | 0.8739 |
| ACONTa | 0.0000 | 0.0 | 0.0 | 0.0000 | 0.0000 |
| ACONTb | 0.0000 | 0.0 | 0.0 | 0.0000 | 0.0000 |
| ACt2r | 0.8739 | 0.0 | 0.0 | 0.8739 | 0.8739 |
| ADK1 | 0.8739 | 0.0 | 0.0 | 0.8739 | 0.8739 |
Phenotype Phase Plane¶
Phenotype phase planes will show distinct phases of optimal growth with different use of two different substrates. For more information, see Edwards et al.
Cobrapy supports calculating and plotting (using matplotlib) these phenotype phase planes. Here, we will make one for the “textbook” E. coli core model.
In [1]:
%matplotlib inline
from IPython.display import set_matplotlib_formats
set_matplotlib_formats('png', 'pdf')
from time import time
import cobra.test
from cobra.flux_analysis import calculate_phenotype_phase_plane
model = cobra.test.create_test_model("textbook")
We want to make a phenotype phase plane to evaluate uptakes of Glucose and Oxygen.
In [2]:
data = calculate_phenotype_phase_plane(
model, "EX_glc__D_e", "EX_o2_e")
data.plot_matplotlib();

If palettable is installed, other color schemes can be used as well
In [3]:
data.plot_matplotlib("Pastel1")
data.plot_matplotlib("Dark2");


The number of points which are plotted in each dimension can also be changed
In [4]:
data = calculate_phenotype_phase_plane(
model, "EX_glc__D_e", "EX_o2_e",
reaction1_npoints=20, reaction2_npoints=20)
data.plot_matplotlib();

The code can also use multiple processes to speed up calculations
In [5]:
start_time = time()
calculate_phenotype_phase_plane(
model, "EX_glc__D_e", "EX_o2_e",
reaction1_npoints=100, reaction2_npoints=100,
n_processes=1)
print("took %.2f seconds with 1 process" % (time() - start_time))
start_time = time()
calculate_phenotype_phase_plane(
model, "EX_glc__D_e", "EX_o2_e",
reaction1_npoints=100, reaction2_npoints=100,
n_processes=4)
print("took %.2f seconds with 4 process" % (time() - start_time))
took 0.44 seconds with 1 process
took 0.25 seconds with 4 process
Mixed-Integer Linear Programming¶
Ice Cream¶
This example was originally contributed by Joshua Lerman.
An ice cream stand sells cones and popsicles. It wants to maximize its profit, but is subject to a budget.
We can write this problem as a linear program:
max cone \(\cdot\) cone_margin + popsicle \(\cdot\) popsicle margin
subject to
cone \(\cdot\) cone_cost + popsicle \(\cdot\) popsicle_cost \(\le\) budget
In [1]:
cone_selling_price = 7.
cone_production_cost = 3.
popsicle_selling_price = 2.
popsicle_production_cost = 1.
starting_budget = 100.
This problem can be written as a cobra.Model
In [2]:
from cobra import Model, Metabolite, Reaction
cone = Reaction("cone")
popsicle = Reaction("popsicle")
# constrainted to a budget
budget = Metabolite("budget")
budget._constraint_sense = "L"
budget._bound = starting_budget
cone.add_metabolites({budget: cone_production_cost})
popsicle.add_metabolites({budget: popsicle_production_cost})
# objective coefficient is the profit to be made from each unit
cone.objective_coefficient = \
cone_selling_price - cone_production_cost
popsicle.objective_coefficient = \
popsicle_selling_price - popsicle_production_cost
m = Model("lerman_ice_cream_co")
m.add_reactions((cone, popsicle))
m.optimize().x_dict
Out[2]:
{'cone': 33.333333333333336, 'popsicle': 0.0}
In reality, cones and popsicles can only be sold in integer amounts. We can use the variable kind attribute of a cobra.Reaction to enforce this.
In [3]:
cone.variable_kind = "integer"
popsicle.variable_kind = "integer"
m.optimize().x_dict
Out[3]:
{'cone': 33.0, 'popsicle': 1.0}
Now the model makes both popsicles and cones.
Restaurant Order¶
To tackle the less immediately obvious problem from the following XKCD comic:
In [4]:
from IPython.display import Image
Image(url=r"http://imgs.xkcd.com/comics/np_complete.png")
Out[4]:
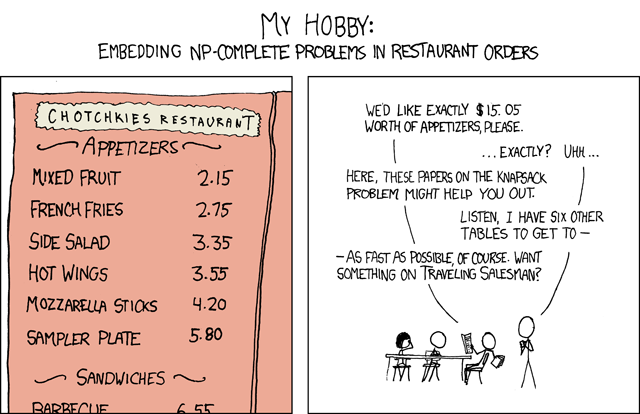
We want a solution satisfying the following constraints:
\(\left(\begin{matrix}2.15&2.75&3.35&3.55&4.20&5.80\end{matrix}\right) \cdot \vec v = 15.05\)
\(\vec v_i \ge 0\)
\(\vec v_i \in \mathbb{Z}\)
This problem can be written as a COBRA model as well.
In [5]:
total_cost = Metabolite("constraint")
total_cost._bound = 15.05
costs = {"mixed_fruit": 2.15, "french_fries": 2.75,
"side_salad": 3.35, "hot_wings": 3.55,
"mozarella_sticks": 4.20, "sampler_plate": 5.80}
m = Model("appetizers")
for item, cost in costs.items():
r = Reaction(item)
r.add_metabolites({total_cost: cost})
r.variable_kind = "integer"
m.add_reaction(r)
# To add to the problem, suppose we want to
# eat as little mixed fruit as possible.
m.reactions.mixed_fruit.objective_coefficient = 1
m.optimize(objective_sense="minimize").x_dict
Out[5]:
{'french_fries': 0.0,
'hot_wings': 2.0,
'mixed_fruit': 1.0,
'mozarella_sticks': 0.0,
'sampler_plate': 1.0,
'side_salad': 0.0}
There is another solution to this problem, which would have been obtained if we had maximized for mixed fruit instead of minimizing.
In [6]:
m.optimize(objective_sense="maximize").x_dict
Out[6]:
{'french_fries': 0.0,
'hot_wings': 0.0,
'mixed_fruit': 7.0,
'mozarella_sticks': 0.0,
'sampler_plate': 0.0,
'side_salad': 0.0}
Boolean Indicators¶
To give a COBRA-related example, we can create boolean variables as integers, which can serve as indicators for a reaction being active in a model. For a reaction flux \(v\) with lower bound -1000 and upper bound 1000, we can create a binary variable \(b\) with the following constraints:
\(b \in \{0, 1\}\)
\(-1000 \cdot b \le v \le 1000 \cdot b\)
To introduce the above constraints into a cobra model, we can rewrite them as follows
\(v \le b \cdot 1000 \Rightarrow v- 1000\cdot b \le 0\)
\(-1000 \cdot b \le v \Rightarrow v + 1000\cdot b \ge 0\)
In [7]:
import cobra.test
model = cobra.test.create_test_model("textbook")
# an indicator for pgi
pgi = model.reactions.get_by_id("PGI")
# make a boolean variable
pgi_indicator = Reaction("indicator_PGI")
pgi_indicator.lower_bound = 0
pgi_indicator.upper_bound = 1
pgi_indicator.variable_kind = "integer"
# create constraint for v - 1000 b <= 0
pgi_plus = Metabolite("PGI_plus")
pgi_plus._constraint_sense = "L"
# create constraint for v + 1000 b >= 0
pgi_minus = Metabolite("PGI_minus")
pgi_minus._constraint_sense = "G"
pgi_indicator.add_metabolites({pgi_plus: -1000,
pgi_minus: 1000})
pgi.add_metabolites({pgi_plus: 1, pgi_minus: 1})
model.add_reaction(pgi_indicator)
# an indicator for zwf
zwf = model.reactions.get_by_id("G6PDH2r")
zwf_indicator = Reaction("indicator_ZWF")
zwf_indicator.lower_bound = 0
zwf_indicator.upper_bound = 1
zwf_indicator.variable_kind = "integer"
# create constraint for v - 1000 b <= 0
zwf_plus = Metabolite("ZWF_plus")
zwf_plus._constraint_sense = "L"
# create constraint for v + 1000 b >= 0
zwf_minus = Metabolite("ZWF_minus")
zwf_minus._constraint_sense = "G"
zwf_indicator.add_metabolites({zwf_plus: -1000,
zwf_minus: 1000})
zwf.add_metabolites({zwf_plus: 1, zwf_minus: 1})
# add the indicator reactions to the model
model.add_reaction(zwf_indicator)
In a model with both these reactions active, the indicators will also be active
In [8]:
solution = model.optimize()
print("PGI indicator = %d" % solution.x_dict["indicator_PGI"])
print("ZWF indicator = %d" % solution.x_dict["indicator_ZWF"])
print("PGI flux = %.2f" % solution.x_dict["PGI"])
print("ZWF flux = %.2f" % solution.x_dict["G6PDH2r"])
PGI indicator = 1
ZWF indicator = 1
PGI flux = 4.86
ZWF flux = 4.96
Because these boolean indicators are in the model, additional constraints can be applied on them. For example, we can prevent both reactions from being active at the same time by adding the following constraint:
\(b_\text{pgi} + b_\text{zwf} = 1\)
In [9]:
or_constraint = Metabolite("or")
or_constraint._bound = 1
zwf_indicator.add_metabolites({or_constraint: 1})
pgi_indicator.add_metabolites({or_constraint: 1})
solution = model.optimize()
print("PGI indicator = %d" % solution.x_dict["indicator_PGI"])
print("ZWF indicator = %d" % solution.x_dict["indicator_ZWF"])
print("PGI flux = %.2f" % solution.x_dict["PGI"])
print("ZWF flux = %.2f" % solution.x_dict["G6PDH2r"])
PGI indicator = 1
ZWF indicator = 0
PGI flux = 9.82
ZWF flux = 0.00
Quadratic Programming¶
Suppose we want to minimize the Euclidean distance of the solution to the origin while subject to linear constraints. This will require a quadratic objective function. Consider this example problem:
min \(\frac{1}{2}\left(x^2 + y^2 \right)\)
subject to
\(x + y = 2\)
\(x \ge 0\)
\(y \ge 0\)
This problem can be visualized graphically:
In [1]:
%matplotlib inline
import plot_helper
plot_helper.plot_qp1()

The objective can be rewritten as \(\frac{1}{2} v^T \cdot \mathbf Q \cdot v\), where \(v = \left(\begin{matrix} x \\ y\end{matrix} \right)\) and \(\mathbf Q = \left(\begin{matrix} 1 & 0\\ 0 & 1 \end{matrix}\right)\)
The matrix \(\mathbf Q\) can be passed into a cobra model as the quadratic objective.
In [2]:
import scipy
from cobra import Reaction, Metabolite, Model, solvers
The quadratic objective \(\mathbf Q\) should be formatted as a scipy sparse matrix.
In [3]:
Q = scipy.sparse.eye(2).todok()
Q
Out[3]:
<2x2 sparse matrix of type '<class 'numpy.float64'>'
with 2 stored elements in Dictionary Of Keys format>
In this case, the quadratic objective is simply the identity matrix
In [4]:
Q.todense()
Out[4]:
matrix([[ 1., 0.],
[ 0., 1.]])
We need to use a solver that supports quadratic programming, such as gurobi or cplex. If a solver which supports quadratic programming is installed, this function will return its name.
In [5]:
print(solvers.get_solver_name(qp=True))
cplex
In [6]:
c = Metabolite("c")
c._bound = 2
x = Reaction("x")
y = Reaction("y")
x.add_metabolites({c: 1})
y.add_metabolites({c: 1})
m = Model()
m.add_reactions([x, y])
sol = m.optimize(quadratic_component=Q, objective_sense="minimize")
sol.x_dict
Out[6]:
{'x': 1.0, 'y': 1.0}
Suppose we change the problem to have a mixed linear and quadratic objective.
min \(\frac{1}{2}\left(x^2 + y^2 \right) - y\)
subject to
\(x + y = 2\)
\(x \ge 0\)
\(y \ge 0\)
Graphically, this would be
In [7]:
plot_helper.plot_qp2()

QP solvers in cobrapy will combine linear and quadratic coefficients. The linear portion will be obtained from the same objective_coefficient attribute used with LP’s.
In [8]:
y.objective_coefficient = -1
sol = m.optimize(quadratic_component=Q, objective_sense="minimize")
sol.x_dict
Out[8]:
{'x': 0.5, 'y': 1.5}
Loopless FBA¶
The goal of this procedure is identification of a thermodynamically consistent flux state without loops, as implied by the name.
Usually, the model has the following constraints.
However, this will allow for thermodynamically infeasible loops (referred to as type 3 loops) to occur, where flux flows around a cycle without any net change of metabolites. For most cases, this is not a major issue, as solutions with these loops can usually be converted to equivalent solutions without them. However, if a flux state is desired which does not exhibit any of these loops, loopless FBA can be used. The formulation used here is modified from Schellenberger et al.
We can make the model irreversible, so that all reactions will satisfy
We will add in boolean indicators as well, such that
We also want to ensure that an entry in the row space of S also exists with negative values wherever v is nonzero. In this expression, \(1-i\) acts as a not to indicate inactivity of a reaction.
We will construct an LP integrating both constraints.
Note that these extra constraints are not applied to boundary reactions which bring metabolites in and out of the system.
In [1]:
%matplotlib inline
import plot_helper
import cobra.test
from cobra import Reaction, Metabolite, Model
from cobra.flux_analysis.loopless import construct_loopless_model
from cobra.flux_analysis import optimize_minimal_flux
from cobra.solvers import get_solver_name
We will demonstrate with a toy model which has a simple loop cycling A -> B -> C -> A, with A allowed to enter the system and C allowed to leave. A graphical view of the system is drawn below:
In [2]:
plot_helper.plot_loop()

In [3]:
test_model = Model()
test_model.add_metabolites([Metabolite(i) for i in "ABC"])
test_model.add_reactions([Reaction(i) for i in
["EX_A", "DM_C", "v1", "v2", "v3"]])
test_model.reactions.EX_A.add_metabolites({"A": 1})
test_model.reactions.DM_C.add_metabolites({"C": -1})
test_model.reactions.DM_C.objective_coefficient = 1
test_model.reactions.v1.add_metabolites({"A": -1, "B": 1})
test_model.reactions.v2.add_metabolites({"B": -1, "C": 1})
test_model.reactions.v3.add_metabolites({"C": -1, "A": 1})
While this model contains a loop, a flux state exists which has no flux through reaction v3, and is identified by loopless FBA.
In [4]:
solution = construct_loopless_model(test_model).optimize()
print("loopless solution: status = " + solution.status)
print("loopless solution: v3 = %.1f" % solution.x_dict["v3"])
loopless solution: status = optimal
loopless solution: v3 = 0.0
If there there is no forced flux through a loopless reaction, parsimonious FBA will also have no flux through the loop.
In [5]:
solution = optimize_minimal_flux(test_model)
print("parsimonious solution: status = " + solution.status)
print("parsimonious solution: v3 = %.1f" % solution.x_dict["v3"])
parsimonious solution: status = optimal
parsimonious solution: v3 = 0.0
However, if flux is forced through v3, then there is no longer a feasible loopless solution, but the parsimonious solution will still exist.
In [6]:
test_model.reactions.v3.lower_bound = 1
solution = construct_loopless_model(test_model).optimize()
print("loopless solution: status = " + solution.status)
loopless solution: status = infeasible
In [7]:
solution = optimize_minimal_flux(test_model)
print("parsimonious solution: status = " + solution.status)
print("parsimonious solution: v3 = %.1f" % solution.x_dict["v3"])
parsimonious solution: status = optimal
parsimonious solution: v3 = 1.0
Loopless FBA is also possible on genome scale models, but it requires a capable MILP solver. If one is installed, cobrapy can detect it automatically using the get_solver_name function
In [8]:
mip_solver = get_solver_name(mip=True)
print(mip_solver)
cplex
In [9]:
salmonella = cobra.test.create_test_model("salmonella")
construct_loopless_model(salmonella).optimize(solver=mip_solver)
Out[9]:
<Solution 0.38 at 0x7f9285d7ffd0>
In [10]:
ecoli = cobra.test.create_test_model("ecoli")
construct_loopless_model(ecoli).optimize(solver=mip_solver)
Out[10]:
<Solution 0.98 at 0x7f9285c89470>
Gapfillling¶
GrowMatch and SMILEY are gap-filling algorithms, which try to to make the minimal number of changes to a model and allow it to simulate growth. For more information, see Kumar et al.. Please note that these algorithms are Mixed-Integer Linear Programs, which need solvers such as gurobi or cplex to function correctly.
In [1]:
import cobra.test
model = cobra.test.create_test_model("salmonella")
In this model D-Fructose-6-phosphate is an essential metabolite. We will remove all the reactions using it, and at them to a separate model.
In [2]:
# remove some reactions and add them to the universal reactions
Universal = cobra.Model("Universal_Reactions")
for i in [i.id for i in model.metabolites.f6p_c.reactions]:
reaction = model.reactions.get_by_id(i)
Universal.add_reaction(reaction.copy())
reaction.remove_from_model()
Now, because of these gaps, the model won’t grow.
In [3]:
model.optimize().f
Out[3]:
2.821531499799383e-12
GrowMatch¶
We will use GrowMatch to add back the minimal number of reactions from this set of “universal” reactions (in this case just the ones we removed) to allow it to grow.
In [4]:
r = cobra.flux_analysis.growMatch(model, Universal)
for e in r[0]:
print(e.id)
GF6PTA
FBP
MAN6PI_reverse
TKT2_reverse
PGI_reverse
We can obtain multiple possible reaction sets by having the algorithm go through multiple iterations.
In [5]:
result = cobra.flux_analysis.growMatch(model, Universal,
iterations=4)
for i, entries in enumerate(result):
print("---- Run %d ----" % (i + 1))
for e in entries:
print(e.id)
---- Run 1 ----
GF6PTA
FBP
MAN6PI_reverse
TKT2_reverse
PGI_reverse
---- Run 2 ----
F6PP
GF6PTA
TALA
MAN6PI_reverse
F6PA_reverse
---- Run 3 ----
GF6PTA
MAN6PI_reverse
TKT2_reverse
F6PA_reverse
PGI_reverse
---- Run 4 ----
F6PP
GF6PTA
FBP
TALA
MAN6PI_reverse
SMILEY¶
SMILEY is very similar to growMatch, only instead of setting growth as the objective, it sets production of a specific metabolite
In [6]:
r = cobra.flux_analysis.gapfilling.SMILEY(model, "ac_e",
Universal)
for e in r[0]:
print(e.id)
GF6PTA
MAN6PI_reverse
TKT2_reverse
F6PA_reverse
PGI_reverse
Solver Interface¶
Each cobrapy solver must expose the following API. The solvers all will have their own distinct LP object types, but each can be manipulated by these functions. This API can be used directly when implementing algorithms efficiently on linear programs because it has 2 primary benefits:
- Avoid the overhead of creating and destroying LP’s for each operation
- Many solver objects preserve the basis between subsequent LP’s, making each subsequent LP solve faster
We will walk though the API with the cglpk solver, which links the cobrapy solver API with GLPK‘s C API.
In [1]:
import cobra.test
model = cobra.test.create_test_model("textbook")
solver = cobra.solvers.cglpk
Attributes and functions¶
Each solver has some attributes:
solver_name¶
The name of the solver. This is the name which will be used to select the solver in cobrapy functions.
In [2]:
solver.solver_name
Out[2]:
'cglpk'
In [3]:
model.optimize(solver="cglpk")
Out[3]:
<Solution 0.87 at 0x7fd42ad90c18>
_SUPPORTS_MILP¶
The presence of this attribute tells cobrapy that the solver supports mixed-integer linear programming
In [4]:
solver._SUPPORTS_MILP
Out[4]:
True
solve¶
Model.optimize is a wrapper for each solver’s solve function. It takes in a cobra model and returns a solution
In [5]:
solver.solve(model)
Out[5]:
<Solution 0.87 at 0x7fd42ad90908>
create_problem¶
This creates the LP object for the solver.
In [6]:
lp = solver.create_problem(model, objective_sense="maximize")
lp
Out[6]:
<cobra.solvers.cglpk.GLP at 0x3e846e8>
solve_problem¶
Solve the LP object and return the solution status
In [7]:
solver.solve_problem(lp)
Out[7]:
'optimal'
format_solution¶
Extract a cobra.Solution object from a solved LP object
In [8]:
solver.format_solution(lp, model)
Out[8]:
<Solution 0.87 at 0x7fd42ad90668>
get_objective_value¶
Extract the objective value from a solved LP object
In [9]:
solver.get_objective_value(lp)
Out[9]:
0.8739215069684909
get_status¶
Get the solution status of a solved LP object
In [10]:
solver.get_status(lp)
Out[10]:
'optimal'
change_variable_objective¶
change the objective coefficient a reaction at a particular index. This does not change any of the other objectives which have already been set. This example will double and then revert the biomass coefficient.
In [11]:
model.reactions.index("Biomass_Ecoli_core")
Out[11]:
12
In [12]:
solver.change_variable_objective(lp, 12, 2)
solver.solve_problem(lp)
solver.get_objective_value(lp)
Out[12]:
1.7478430139369818
In [13]:
solver.change_variable_objective(lp, 12, 1)
solver.solve_problem(lp)
solver.get_objective_value(lp)
Out[13]:
0.8739215069684909
change variable_bounds¶
change the lower and upper bounds of a reaction at a particular index. This example will set the lower bound of the biomass to an infeasible value, then revert it.
In [14]:
solver.change_variable_bounds(lp, 12, 1000, 1000)
solver.solve_problem(lp)
Out[14]:
'infeasible'
In [15]:
solver.change_variable_bounds(lp, 12, 0, 1000)
solver.solve_problem(lp)
Out[15]:
'optimal'
change_coefficient¶
Change a coefficient in the stoichiometric matrix. In this example, we will set the entry for ADP in the ATMP reaction to in infeasible value, then reset it.
In [16]:
model.metabolites.index("atp_c")
Out[16]:
16
In [17]:
model.reactions.index("ATPM")
Out[17]:
10
In [18]:
solver.change_coefficient(lp, 16, 10, -10)
solver.solve_problem(lp)
Out[18]:
'infeasible'
In [19]:
solver.change_coefficient(lp, 16, 10, -1)
solver.solve_problem(lp)
Out[19]:
'optimal'
set_parameter¶
Set a solver parameter. Each solver will have its own particular set of unique paramters. However, some have unified names. For example, all solvers should accept “tolerance_feasibility.”
In [20]:
solver.set_parameter(lp, "tolerance_feasibility", 1e-9)
In [21]:
solver.set_parameter(lp, "objective_sense", "minimize")
solver.solve_problem(lp)
solver.get_objective_value(lp)
Out[21]:
0.0
In [22]:
solver.set_parameter(lp, "objective_sense", "maximize")
solver.solve_problem(lp)
solver.get_objective_value(lp)
Out[22]:
0.8739215069684912
Example with FVA¶
Consider flux variability analysis (FVA), which requires maximizing and minimizing every reaction with the original biomass value fixed at its optimal value. If we used the cobra Model API in a naive implementation, we would do the following:
In [23]:
%%time
# work on a copy of the model so the original is not changed
m = model.copy()
# set the lower bound on the objective to be the optimal value
f = m.optimize().f
for objective_reaction, coefficient in m.objective.items():
objective_reaction.lower_bound = coefficient * f
# now maximize and minimze every reaction to find its bounds
fva_result = {}
for r in m.reactions:
m.change_objective(r)
fva_result[r.id] = {
"maximum": m.optimize(objective_sense="maximize").f,
"minimum": m.optimize(objective_sense="minimize").f
}
CPU times: user 171 ms, sys: 0 ns, total: 171 ms
Wall time: 171 ms
Instead, we could use the solver API to do this more efficiently. This is roughly how cobrapy implementes FVA. It keeps uses the same LP object and repeatedly maximizes and minimizes it. This allows the solver to preserve the basis, and is much faster. The speed increase is even more noticeable the larger the model gets.
In [24]:
%%time
# create the LP object
lp = solver.create_problem(model)
# set the lower bound on the objective to be the optimal value
solver.solve_problem(lp)
f = solver.get_objective_value(lp)
for objective_reaction, coefficient in model.objective.items():
objective_index = model.reactions.index(objective_reaction)
# old objective is no longer the objective
solver.change_variable_objective(lp, objective_index, 0.)
solver.change_variable_bounds(
lp, objective_index, f * coefficient,
objective_reaction.upper_bound)
# now maximize and minimze every reaction to find its bounds
fva_result = {}
for index, r in enumerate(model.reactions):
solver.change_variable_objective(lp, index, 1.)
result = {}
solver.solve_problem(lp, objective_sense="maximize")
result["maximum"] = solver.get_objective_value(lp)
solver.solve_problem(lp, objective_sense="minimize")
result["minimum"] = solver.get_objective_value(lp)
solver.change_variable_objective(lp, index, 0.)
fva_result[r.id] = result
CPU times: user 8.28 ms, sys: 25 µs, total: 8.31 ms
Wall time: 8.14 ms
Using the COBRA toolbox with cobrapy¶
This example demonstrates using COBRA toolbox commands in MATLAB from python through pymatbridge.
In [1]:
%load_ext pymatbridge
Starting MATLAB on ZMQ socket ipc:///tmp/pymatbridge-57ff5429-02d9-4e1a-8ed0-44e391fb0df7
Send 'exit' command to kill the server
....MATLAB started and connected!
In [2]:
import cobra.test
m = cobra.test.create_test_model("textbook")
The model_to_pymatbridge function will send the model to the workspace with the given variable name.
In [3]:
from cobra.io.mat import model_to_pymatbridge
model_to_pymatbridge(m, variable_name="model")
Now in the MATLAB workspace, the variable name ‘model’ holds a COBRA toolbox struct encoding the model.
In [4]:
%%matlab
model
model =
rev: [95x1 double]
metNames: {72x1 cell}
b: [72x1 double]
metCharge: [72x1 double]
c: [95x1 double]
csense: [72x1 char]
genes: {137x1 cell}
metFormulas: {72x1 cell}
rxns: {95x1 cell}
grRules: {95x1 cell}
rxnNames: {95x1 cell}
description: [11x1 char]
S: [72x95 double]
ub: [95x1 double]
lb: [95x1 double]
mets: {72x1 cell}
subSystems: {95x1 cell}
First, we have to initialize the COBRA toolbox in MATLAB.
In [5]:
%%matlab --silent
warning('off'); % this works around a pymatbridge bug
addpath(genpath('~/cobratoolbox/'));
initCobraToolbox();
Commands from the COBRA toolbox can now be run on the model
In [6]:
%%matlab
optimizeCbModel(model)
ans =
x: [95x1 double]
f: 0.8739
y: [71x1 double]
w: [95x1 double]
stat: 1
origStat: 5
solver: 'glpk'
time: 3.2911
FBA in the COBRA toolbox should give the same result as cobrapy (but maybe just a little bit slower :))
In [7]:
%time
m.optimize().f
CPU times: user 0 ns, sys: 0 ns, total: 0 ns
Wall time: 5.48 µs
Out[7]:
0.8739215069684909
FAQ¶
This document will address frequently asked questions not addressed in other pages of the documentation.
How do I install cobrapy?¶
Please see the INSTALL.rst file.
How do I cite cobrapy?¶
Please cite the 2013 publication: 10.1186/1752-0509-7-74
How do I rename reactions or metabolites?¶
TL;DR Use Model.repair afterwards
When renaming metabolites or reactions, there are issues because cobra indexes based off of ID’s, which can cause errors. For example:
In [1]:
from __future__ import print_function
import cobra.test
model = cobra.test.create_test_model()
for metabolite in model.metabolites:
metabolite.id = "test_" + metabolite.id
try:
model.metabolites.get_by_id(model.metabolites[0].id)
except KeyError as e:
print(repr(e))
KeyError('test_dcaACP_c',)
The Model.repair function will rebuild the necessary indexes
In [2]:
model.repair()
model.metabolites.get_by_id(model.metabolites[0].id)
Out[2]:
<Metabolite test_dcaACP_c at 0x7f90c2b97978>
How do I delete a gene?¶
That depends on what precisely you mean by delete a gene.
If you want to simulate the model with a gene knockout, use the cobra.maniupulation.delete_model_genes function. The effects of this function are reversed by cobra.manipulation.undelete_model_genes.
In [3]:
model = cobra.test.create_test_model()
PGI = model.reactions.get_by_id("PGI")
print("bounds before knockout:", (PGI.lower_bound, PGI.upper_bound))
cobra.manipulation.delete_model_genes(model, ["STM4221"])
print("bounds after knockouts", (PGI.lower_bound, PGI.upper_bound))
bounds before knockout: (-1000.0, 1000.0)
bounds after knockouts (0.0, 0.0)
If you want to actually remove all traces of a gene from a model, this is more difficult because this will require changing all the gene_reaction_rule strings for reactions involving the gene.
How do I change the reversibility of a Reaction?¶
Reaction.reversibility is a property in cobra which is computed when it is requested from the lower and upper bounds.
In [4]:
model = cobra.test.create_test_model()
model.reactions.get_by_id("PGI").reversibility
Out[4]:
True
Trying to set it directly will result in an error or warning:
In [5]:
try:
model.reactions.get_by_id("PGI").reversibility = False
except Exception as e:
print(repr(e))
cobra/core/Reaction.py:192 UserWarning: Setting reaction reversibility is ignored
The way to change the reversibility is to change the bounds to make the reaction irreversible.
In [6]:
model.reactions.get_by_id("PGI").lower_bound = 10
model.reactions.get_by_id("PGI").reversibility
Out[6]:
False
How do I generate an LP file from a COBRA model?¶
While the cobrapy does not include python code to support this feature directly, many of the bundled solvers have this capability. Create the problem with one of these solvers, and use its appropriate function.
Please note that unlike the LP file format, the MPS file format does not specify objective direction and is always a minimzation. Some (but not all) solvers will rewrite the maximization as a minimzation.
In [7]:
model = cobra.test.create_test_model()
# glpk through cglpk
glp = cobra.solvers.cglpk.create_problem(model)
glp.write("test.lp")
glp.write("test.mps") # will not rewrite objective
# gurobi
gurobi_problem = cobra.solvers.gurobi_solver.create_problem(model)
gurobi_problem.write("test.lp")
gurobi_problem.write("test.mps") # rewrites objective
# cplex
cplex_problem = cobra.solvers.cplex_solver.create_problem(model)
cplex_problem.write("test.lp")
cplex_problem.write("test.mps") # rewrites objective
---------------------------------------------------------------------------
AttributeError Traceback (most recent call last)
<ipython-input-7-76bbced9b9d6> in <module>()
5 glp.write("test.mps") # will not rewrite objective
6 # gurobi
----> 7 gurobi_problem = cobra.solvers.gurobi_solver.create_problem(model)
8 gurobi_problem.write("test.lp")
9 gurobi_problem.write("test.mps") # rewrites objective
AttributeError: 'module' object has no attribute 'gurobi_solver'
How do I visualize my flux solutions?¶
cobrapy works well with the escher package, which is well suited to this purpose. Consult the escher documentation for examples.
cobra package¶
Subpackages¶
cobra.core package¶
Submodules¶
cobra.core.ArrayBasedModel module¶
-
class
cobra.core.ArrayBasedModel.ArrayBasedModel(description=None, deepcopy_model=False, matrix_type='scipy.lil_matrix')[source]¶ Bases:
cobra.core.Model.ModelArrayBasedModel is a class that adds arrays and vectors to a cobra.Model to make it easier to perform linear algebra operations.
-
S¶ Stoichiometric matrix of the model
This will be formatted as either
lil_matrixordok_matrix
-
add_metabolites(metabolite_list, expand_stoichiometric_matrix=True)[source]¶ Will add a list of metabolites to the the object, if they do not exist and then expand the stochiometric matrix
metabolite_list: A list of
Metaboliteobjectsexpand_stoichimetric_matrix: Boolean. If True and self.S is not None then it will add rows to self.S. self.S must be created after adding reactions and metabolites to self before it can be expanded. Trying to expand self.S when self only contains metabolites is ludacris.
-
add_reactions(reaction_list, update_matrices=True)[source]¶ Will add a cobra.Reaction object to the model, if reaction.id is not in self.reactions.
reaction_list: A
Reactionobject or a list of themupdate_matrices: Boolean. If true populate / update matrices S, lower_bounds, upper_bounds, .... Note this is slow to run for very large models and using this option with repeated calls will degrade performance. Better to call self.update() after adding all reactions.
If the stoichiometric matrix is initially empty then initialize a 1x1 sparse matrix and add more rows as needed in the self.add_metabolites function
-
b¶ bounds for metabolites as
numpy.ndarray
-
constraint_sense¶
-
copy()[source]¶ Provides a partial ‘deepcopy’ of the Model. All of the Metabolite, Gene, and Reaction objects are created anew but in a faster fashion than deepcopy
-
lower_bounds¶
-
objective_coefficients¶
-
remove_reactions(reactions, update_matrices=True, **kwargs)[source]¶ remove reactions from the model
See
cobra.core.Model.Model.remove_reactions()- update_matrices: Boolean
- If true populate / update matrices S, lower_bounds, upper_bounds. Note that this is slow to run for very large models, and using this option with repeated calls will degrade performance.
-
upper_bounds¶
-
cobra.core.DictList module¶
-
class
cobra.core.DictList.DictList(*args)[source]¶ Bases:
listA combined dict and list
This object behaves like a list, but has the O(1) speed benefits of a dict when looking up elements by their id.
-
__add__(other)[source]¶ x.__add__(y) <==> x + y
- other: iterable
- other must contain only unique id’s which do not intersect with self
-
__contains__(object)[source]¶ DictList.__contains__(object) <==> object in DictList
object: str or
Object
-
__getstate__()[source]¶ gets internal state
This is only provided for backwards compatibilty so older versions of cobrapy can load pickles generated with cobrapy. In reality, the “_dict” state is ignored when loading a pickle
-
__iadd__(other)[source]¶ x.__iadd__(y) <==> x += y
- other: iterable
- other must contain only unique id’s whcih do not intersect with self
-
__setstate__(state)[source]¶ sets internal state
Ignore the passed in state and recalculate it. This is only for compatibility with older pickles which did not correctly specify the initialization class
-
query(search_function, attribute='id')[source]¶ query the list
- search_function: used to select which objects to return
- a string, in which case any object.attribute containing the string will be returned
- a compiled regular expression
- a function which takes one argument and returns True for desired values
- attribute: the attribute to be searched for (default is ‘id’).
- If this is None, the object itself is used.
returns: a list of objects which match the query
-
cobra.core.Formula module¶
-
class
cobra.core.Formula.Formula(formula=None)[source]¶ Bases:
cobra.core.Object.ObjectDescribes a Chemical Formula
A legal formula string contains only letters and numbers.
-
__add__(other_formula)[source]¶ Combine two molecular formulas.
other_formula: cobra.Formula or str of a chemical Formula.
-
weight¶ Calculate the formula weight
-
cobra.core.Gene module¶
-
class
cobra.core.Gene.GPRCleaner[source]¶ Bases:
ast.NodeTransformerParses compiled ast of a gene_reaction_rule and identifies genes
Parts of the tree are rewritten to allow periods in gene ID’s and bitwise boolean operations
-
class
cobra.core.Gene.Gene(id=None, name='', functional=True)[source]¶ Bases:
cobra.core.Species.Species-
remove_from_model(model=None, make_dependent_reactions_nonfunctional=True)[source]¶ Removes the association
make_dependent_reactions_nonfunctional: Boolean. If True then replace the gene with ‘False’ in the gene association, else replace the gene with ‘True’
Deprecated since version 0.4: Use cobra.manipulation.delete_model_genes to simulate knockouts and cobra.manipulation.remove_genes to remove genes from the model.
-
-
cobra.core.Gene.ast2str(expr, level=0, names=None)[source]¶ convert compiled ast to gene_reaction_rule str
expr: str of a gene reaction rule
level: internal use only
- names: optional dict of {Gene.id: Gene.name}
- Use this to get a rule str which uses names instead. This should be done for display purposes only. All gene_reaction_rule strings which are computed with should use the id.
cobra.core.Metabolite module¶
-
class
cobra.core.Metabolite.Metabolite(id=None, formula=None, name='', charge=None, compartment=None)[source]¶ Bases:
cobra.core.Species.SpeciesMetabolite is a class for holding information regarding a metabolite in a cobra.Reaction object.
-
elements¶
-
formula_weight¶ Calculate the formula weight
-
remove_from_model(method='subtractive', **kwargs)[source]¶ Removes the association from self.model
- method: ‘subtractive’ or ‘destructive’.
- If ‘subtractive’ then the metabolite is removed from all associated reactions. If ‘destructive’ then all associated reactions are removed from the Model.
-
summary(**kwargs)[source]¶ Print a summary of the reactions which produce and consume this metabolite. This method requires the model for which this metabolite is a part to be solved.
- threshold: float
- a value below which to ignore reaction fluxes
- fva: float (0->1), or None
- Whether or not to include flux variability analysis in the output. If given, fva should be a float between 0 and 1, representing the fraction of the optimum objective to be searched.
- floatfmt: string
- format method for floats, passed to tabulate. Default is ‘.3g’.
-
y¶ The shadow price for the metabolite in the most recent solution
Shadow prices are computed from the dual values of the bounds in the solution.
-
cobra.core.Model module¶
-
class
cobra.core.Model.Model(id_or_model=None, name=None)[source]¶ Bases:
cobra.core.Object.ObjectMetabolic Model
Refers to Metabolite, Reaction, and Gene Objects.
-
__add__(other_model)[source]¶ Adds two models. +
The issue of reactions being able to exists in multiple Models now arises, the same for metabolites and such. This might be a little difficult as a reaction with the same name / id in two models might have different coefficients for their metabolites due to pH and whatnot making them different reactions.
-
__iadd__(other_model)[source]¶ Adds a Model to this model +=
The issue of reactions being able to exists in multiple Models now arises, the same for metabolites and such. This might be a little difficult as a reaction with the same name / id in two models might have different coefficients for their metabolites due to pH and whatnot making them different reactions.
-
add_metabolites(metabolite_list)[source]¶ Will add a list of metabolites to the the object, if they do not exist and then expand the stochiometric matrix
metabolite_list: A list of
Metaboliteobjects
-
add_reaction(reaction)[source]¶ Will add a cobra.Reaction object to the model, if reaction.id is not in self.reactions.
reaction: A
Reactionobject
-
add_reactions(reaction_list)[source]¶ Will add a cobra.Reaction object to the model, if reaction.id is not in self.reactions.
reaction_list: A list of
Reactionobjects
-
copy()[source]¶ Provides a partial ‘deepcopy’ of the Model. All of the Metabolite, Gene, and Reaction objects are created anew but in a faster fashion than deepcopy
-
description¶
-
objective¶
-
optimize(objective_sense='maximize', **kwargs)[source]¶ Optimize model using flux balance analysis
objective_sense: ‘maximize’ or ‘minimize’
solver: ‘glpk’, ‘cglpk’, ‘gurobi’, ‘cplex’ or None
- quadratic_component: None or
scipy.sparse.dok_matrix The dimensions should be (n, n) where n is the number of reactions.
This sets the quadratic component (Q) of the objective coefficient, adding \(\\frac{1}{2} v^T \cdot Q \cdot v\) to the objective.
tolerance_feasibility: Solver tolerance for feasibility.
tolerance_markowitz: Solver threshold during pivot
time_limit: Maximum solver time (in seconds)
Note
Only the most commonly used parameters are presented here. Additional parameters for cobra.solvers may be available and specified with the appropriate keyword argument.
- quadratic_component: None or
-
remove_reactions(reactions, delete=True, remove_orphans=False)[source]¶ remove reactions from the model
- reactions: [
Reaction] or [str] - The reactions (or their id’s) to remove
- delete: Boolean
- Whether or not the reactions should be deleted after removal. If the reactions are not deleted, those objects will be recreated with new metabolite and gene objects.
- remove_orphans: Boolean
- Remove orphaned genes and metabolites from the model as well
- reactions: [
-
repair(rebuild_index=True, rebuild_relationships=True)[source]¶ Update all indexes and pointers in a model
-
summary(**kwargs)[source]¶ Print a summary of the input and output fluxes of the model. This method requires the model to have been previously solved.
- threshold: float
- tolerance for determining if a flux is zero (not printed)
- fva: int or None
- Whether or not to calculate and report flux variability in the output summary
- floatfmt: string
- format method for floats, passed to tabulate. Default is ‘.3g’.
-
to_array_based_model(deepcopy_model=False, **kwargs)[source]¶ Makes a
ArrayBasedModelfrom a cobra.Model which may be used to perform linear algebra operations with the stoichiomatric matrix.deepcopy_model: Boolean. If False then the ArrayBasedModel points to the Model
-
cobra.core.Object module¶
cobra.core.Reaction module¶
-
class
cobra.core.Reaction.Reaction(id=None, name='', subsystem='', lower_bound=0.0, upper_bound=1000.0, objective_coefficient=0.0)[source]¶ Bases:
cobra.core.Object.ObjectReaction is a class for holding information regarding a biochemical reaction in a cobra.Model object
-
__add__(other)[source]¶ Add two reactions
The stoichiometry will be the combined stoichiometry of the two reactions, and the gene reaction rule will be both rules combined by an and. All other attributes (i.e. reaction bounds) will match those of the first reaction
-
__setstate__(state)[source]¶ Probably not necessary to set _model as the cobra.Model that contains self sets the _model attribute for all metabolites and genes in the reaction.
However, to increase performance speed we do want to let the metabolite and gene know that they are employed in this reaction
-
add_metabolites(metabolites, combine=True, add_to_container_model=True)[source]¶ Add metabolites and stoichiometric coefficients to the reaction. If the final coefficient for a metabolite is 0 then it is removed from the reaction.
- metabolites: dict
- {str or
Metabolite: coefficient} - combine: Boolean.
- Describes behavior a metabolite already exists in the reaction. True causes the coefficients to be added. False causes the coefficient to be replaced. True and a metabolite already exists in the
- add_to_container_model: Boolean.
- Add the metabolite to the
Modelthe reaction is associated with (i.e. self.model)
-
boundary¶
-
bounds¶ A more convienient bounds structure than seperate upper and lower bounds
-
build_reaction_from_string(reaction_str, verbose=True, fwd_arrow=None, rev_arrow=None, reversible_arrow=None, term_split='+')[source]¶ Builds reaction from reaction equation reaction_str using parser
Takes a string and using the specifications supplied in the optional arguments infers a set of metabolites, metabolite compartments and stoichiometries for the reaction. It also infers the reversibility of the reaction from the reaction arrow.
Parameters: - reaction_str – a string containing a reaction formula (equation)
- verbose – Boolean setting verbosity of function (optional, default=True)
- fwd_arrow – re.compile for forward irreversible reaction arrows (optional, default=_forward_arrow_finder)
- reverse_arrow – re.compile for backward irreversible reaction arrows (optional, default=_reverse_arrow_finder)
- fwd_arrow – re.compile for reversible reaction arrows (optional, default=_reversible_arrow_finder)
- term_split – String dividing individual metabolite entries (optional, default=’+’)
-
build_reaction_string(use_metabolite_names=False)[source]¶ Generate a human readable reaction string
-
check_mass_balance()[source]¶ Compute mass and charge balance for the reaction
returns a dict of {element: amount} for unbalanced elements. “charge” is treated as an element in this dict This should be empty for balanced reactions.
-
delete(remove_orphans=False)[source]¶ Completely delete a reaction
This removes all associations between a reaction the associated model, metabolites and genes (unlike remove_from_model which only dissociates the reaction from the model).
- remove_orphans: Boolean
- Remove orphaned genes and metabolites from the model as well
-
gene_name_reaction_rule¶ Display gene_reaction_rule with names intead.
Do NOT use this string for computation. It is intended to give a representation of the rule using more familiar gene names instead of the often cryptic ids.
-
gene_reaction_rule¶
-
genes¶
-
get_coefficient(metabolite_id)[source]¶ Return the stoichiometric coefficient for a metabolite in the reaction.
metabolite_id: str or
Metabolite
-
get_coefficients(metabolite_ids)[source]¶ Return the stoichiometric coefficients for a list of metabolites in the reaction.
- metabolite_ids: iterable
- Containing str or
Metabolite
-
metabolites¶
-
model¶ returns the model the reaction is a part of
-
pop(metabolite_id)[source]¶ Remove a metabolite from the reaction and return the stoichiometric coefficient.
metabolite_id: str or
Metabolite
-
products¶ Return a list of products for the reaction
-
reactants¶ Return a list of reactants for the reaction.
-
reaction¶ Human readable reaction string
-
remove_from_model(model=None, remove_orphans=False)[source]¶ Removes the reaction from the model while keeping it intact
- remove_orphans: Boolean
- Remove orphaned genes and metabolites from the model as well
model: deprecated argument, must be None
-
reversibility¶ Whether the reaction can proceed in both directions (reversible)
This is computed from the current upper and lower bounds.
-
subtract_metabolites(metabolites, combine=True)[source]¶ This function will ‘subtract’ metabolites from a reaction, which means add the metabolites with -1*coefficient. If the final coefficient for a metabolite is 0 then the metabolite is removed from the reaction.
- metabolites: dict of {
Metabolite: coefficient} - These metabolites will be added to the reaction
Note
A final coefficient < 0 implies a reactant.
- metabolites: dict of {
-
x¶ The flux through the reaction in the most recent solution
Flux values are computed from the primal values of the variables in the solution.
-
cobra.core.Solution module¶
-
class
cobra.core.Solution.Solution(f, x=None, x_dict=None, y=None, y_dict=None, solver=None, the_time=0, status='NA')[source]¶ Bases:
objectStores the solution from optimizing a cobra.Model. This is used to provide a single interface to results from different solvers that store their values in different ways.
f: The objective value
solver: A string indicating which solver package was used.
x: List or Array of the values from the primal.
x_dict: A dictionary of reaction ids that maps to the primal values.
y: List or Array of the values from the dual.
y_dict: A dictionary of reaction ids that maps to the dual values.
cobra.core.Species module¶
-
class
cobra.core.Species.Species(id=None, name=None)[source]¶ Bases:
cobra.core.Object.ObjectSpecies is a class for holding information regarding a chemical Species
-
__getstate__()[source]¶ Remove the references to container reactions when serializing to avoid problems associated with recursion.
-
copy()[source]¶ When copying a reaction, it is necessary to deepcopy the components so the list references aren’t carried over.
Additionally, a copy of a reaction is no longer in a cobra.Model.
This should be fixed with self.__deecopy__ if possible
-
model¶
-
reactions¶
-
Module contents¶
cobra.design package¶
Submodules¶
cobra.design.design_algorithms module¶
-
cobra.design.design_algorithms.dual_problem(model, objective_sense='maximize', integer_vars_to_maintain=[], already_irreversible=False, copy=True, dual_maximum=1000)[source]¶ Return a new model representing the dual of the model.
Make the problem irreversible, then take the dual. Convert the problem:
Maximize (c^T)x subject to Ax <= b, x >= 0
which is something like this in COBRApy:
Maximize sum(objective_coefficient_j * reaction_j for all j) s.t. sum(coefficient_i_j * reaction_j for all j) <= metabolite_bound_i reaction_j <= upper_bound_j reaction_j >= 0to the problem:
Minimize (b^T)w subject to (A^T)w >= c, w >= 0
which is something like this in COBRApy (S matrix is m x n):
Minimize sum( metabolite_bound_i * dual_i for all i ) + sum( upper_bound_j * dual_m+j for all j ) + s.t. sum( coefficient_i_j * dual_i for all i ) + sum( dual_2m+j' for all j' ) >= objective_coefficient_j dual_k >= 0Parameters: - model (
Model) – The COBRA model. - objective_sense (str) – The objective sense of the starting problem, either ‘maximize’ or ‘minimize’. A minimization problems will be converted to a maximization before taking the dual. This function always returns a minimization problem.
- iteger_vars_to_maintain ([str]) – A list of IDs for Boolean integer variables to be maintained in the dual problem. See ‘Maintaining integer variables’ below for more details.
- already_irreversible (bool) – If True, then do not convert the model to irreversible.
- copy (bool) – If True, then make a copy of the model before modifying it. This is not necessary if already_irreversible is True.
- dual_maximum (float or int) – The upper bound for dual variables.
Maintaining integer variables
The argument
integer_vars_to_maintaincan be used to specify certin Boolean integer variables that will be maintained in the dual problem. This makes it possible to join outer and inner problems in a bi-level MILP. The method for maintaining integer variables is described by Tepper and Shlomi, 2010:Tepper N, Shlomi T. Predicting metabolic engineering knockout strategies for chemical production: accounting for competing pathways. Bioinformatics. 2010;26(4):536-43. https://doi.org/10.1093/bioinformatics/btp704.
In COBRApy, this roughly translates to transforming (decision variables p, integer constraints o):
Maximize (c^T)x subject to (A_x)x + (A_y)y <= b, x >= 0 (1) Maximize sum(objective_coefficient_j * reaction_j for all j) s.t. (2) sum(coeff_i_j * reaction_j for all j) + sum(decision_coeff_i_j * decision_var_j for all j) <= metabolite_bound_i (3) reaction_j <= upper_bound_j (4) reaction_j >= 0to the problem:
Minimize (b - (A_y)y)^T w subject to (A_x^T)w >= c, w >= 0
which linearizes to (with auxiliary variables z):
Minimize (b^T)w - { ((A_y)y)^T w with yw --> z } subject to (A_x^T)w >= c, linearization constraints, w >= 0 Linearization constraints: z <= w_max * y, z <= w, z >= w - w_max * (1 - y), z >= 0 (5) Minimize sum( metabolite_bound_i * dual_i for all i ) + sum( upper_bound_j * dual_m+j for all j ) + - sum( decision_coeff_i_j * auxiliary_var_i_j for all combinations i, j ) s.t. (6) - sum( coefficient_i_j * dual_i for all i ) - dual_m+j <= - objective_coefficient_j (7) auxiliary_var_i_j - dual_maximum * decision_var_j <= 0 (8) auxiliary_var_i_j - dual_i <= 0 (9) - auxiliary_var_i_j + dual_i + dual_maximum * decision_var_j <= dual_maximum (10) dual_maximum >= dual_i >= 0 (11) dual_maximum >= dual_m+j >= 0 (12) dual_maximum >= auxiliary_var_i_j >= 0 (13) 1 >= decision_var_j >= 0Zachary King 2015
- model (
-
cobra.design.design_algorithms.run_optknock(optknock_problem, solver=None, tolerance_integer=1e-09, **kwargs)[source]¶ Run the OptKnock problem created with set_up_optknock.
Parameters: Zachary King 2015
-
cobra.design.design_algorithms.set_up_optknock(model, chemical_objective, knockable_reactions, biomass_objective=None, n_knockouts=5, n_knockouts_required=True, dual_maximum=1000, copy=True)[source]¶ Set up the OptKnock problem described by Burgard et al., 2003:
Burgard AP, Pharkya P, Maranas CD. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84(6):647-57. https://doi.org/10.1002/bit.10803.
Parameters: - model (
Model) – A COBRA model. - chemical_objective (str) – The ID of the reaction to maximize in the outer problem.
- knockable_reactions ([str]) – A list of reaction IDs that can be knocked out.
- biomass_objective (str) – The ID of the reaction to maximize in the inner problem. By default, this is the existing objective function in the passed model.
- n_knockouts (int) – The number of knockouts allowable.
- n_knockouts_required (bool) – Require exactly the number of knockouts specified by n_knockouts.
- dual_maximum (float or int) – The upper bound for dual variables.
- copy (bool) – Copy the model before making any modifications.
Zachary King 2015
- model (
Module contents¶
cobra.flux_analysis package¶
Submodules¶
cobra.flux_analysis.deletion_worker module¶
-
class
cobra.flux_analysis.deletion_worker.CobraDeletionMockPool(cobra_model, n_processes=1, solver=None, **kwargs)[source]¶ Bases:
objectMock pool solves LP’s in the same process
-
class
cobra.flux_analysis.deletion_worker.CobraDeletionPool(cobra_model, n_processes=None, solver=None, **kwargs)[source]¶ Bases:
objectA pool of workers for solving deletions
submit jobs to the pool using submit and recieve results using receive_all
-
pids¶
-
cobra.flux_analysis.double_deletion module¶
-
cobra.flux_analysis.double_deletion.double_deletion(cobra_model, element_list_1=None, element_list_2=None, element_type='gene', **kwargs)[source]¶ Wrapper for double_gene_deletion and double_reaction_deletion
Deprecated since version 0.4: Use double_reaction_deletion and double_gene_deletion
-
cobra.flux_analysis.double_deletion.double_gene_deletion(cobra_model, gene_list1=None, gene_list2=None, method='fba', return_frame=False, solver=None, zero_cutoff=1e-12, **kwargs)[source]¶ sequentially knocks out pairs of genes in a model
- cobra_model :
Model - cobra model in which to perform deletions
- gene_list1 : [
Gene:] (or their id’s) - Genes to be deleted. These will be the rows in the result. If not provided, all reactions will be used.
- gene_list1 : [
Gene:] (or their id’s) - Genes to be deleted. These will be the rows in the result. If not provided, reaction_list1 will be used.
- method: “fba” or “moma”
- Procedure used to predict the growth rate
- solver: str for solver name
- This must be a QP-capable solver for MOMA. If left unspecified, a suitable solver will be automatically chosen.
- zero_cutoff: float
- When checking to see if a value is 0, this threshold is used.
- number_of_processes: int for number of processes to use.
- If unspecified, the number of parallel processes to use will be automatically determined. Setting this to 1 explicitly disables used of the multiprocessing library.
Note
multiprocessing is not supported with method=moma
- return_frame: bool
- If true, formats the results as a pandas.Dataframe. Otherwise returns a dict of the form: {“x”: row_labels, “y”: column_labels”, “data”: 2D matrix}
- cobra_model :
-
cobra.flux_analysis.double_deletion.double_reaction_deletion(cobra_model, reaction_list1=None, reaction_list2=None, method='fba', return_frame=False, solver=None, zero_cutoff=1e-12, **kwargs)[source]¶ sequentially knocks out pairs of reactions in a model
- cobra_model :
Model - cobra model in which to perform deletions
- reaction_list1 : [
Reaction:] (or their id’s) - Reactions to be deleted. These will be the rows in the result. If not provided, all reactions will be used.
- reaction_list2 : [
Reaction:] (or their id’s) - Reactions to be deleted. These will be the rows in the result. If not provided, reaction_list1 will be used.
- method: “fba” or “moma”
- Procedure used to predict the growth rate
- solver: str for solver name
- This must be a QP-capable solver for MOMA. If left unspecified, a suitable solver will be automatically chosen.
- zero_cutoff: float
- When checking to see if a value is 0, this threshold is used.
- return_frame: bool
- If true, formats the results as a pandas.Dataframe. Otherwise returns a dict of the form: {“x”: row_labels, “y”: column_labels”, “data”: 2D matrix}
- cobra_model :
-
cobra.flux_analysis.double_deletion.format_results_frame(row_ids, column_ids, matrix, return_frame=False)[source]¶ format results as a pandas.DataFrame if desired/possible
Otherwise returns a dict of {“x”: row_ids, “y”: column_ids”, “data”: result_matrx}
-
cobra.flux_analysis.double_deletion.generate_matrix_indexes(ids1, ids2)[source]¶ map an identifier to an entry in the square result matrix
-
cobra.flux_analysis.double_deletion.yield_upper_tria_indexes(ids1, ids2, id_to_index)[source]¶ gives the necessary indexes in the upper triangle
ids1 and ids2 are lists of the identifiers i.e. gene id’s or reaction indexes to be knocked out. id_to_index maps each identifier to its index in the result matrix.
Note that this does not return indexes for the diagonal. Those have to be computed separately.
cobra.flux_analysis.essentiality module¶
-
cobra.flux_analysis.essentiality.assess_medium_component_essentiality(cobra_model, the_components=None, the_medium=None, medium_compartment='e', solver='glpk', the_condition=None, method='fba')[source]¶ Determines which components in an in silico medium are essential for growth in the context of the remaining components.
cobra_model: A Model object.
the_components: None or a list of external boundary reactions that will be sequentially disabled.
the_medium: Is None, a string, or a dictionary. If a string then the initialize_growth_medium function expects that the_model has an attribute dictionary called media_compositions, which is a dictionary of dictionaries for various medium compositions. Where a medium composition is a dictionary of external boundary reaction ids for the medium components and the external boundary fluxes for each medium component.
medium_compartment: the compartment in which the boundary reactions supplying the medium components exist
NOTE: that these fluxes must be negative because the convention is backwards means something is feed into the system.
solver: ‘glpk’, ‘gurobi’, or ‘cplex’
returns: essentiality_dict: A dictionary providing the maximum growth rate accessible when the respective component is removed from the medium.
cobra.flux_analysis.gapfilling module¶
-
cobra.flux_analysis.gapfilling.SMILEY(model, metabolite_id, Universal, dm_rxns=False, ex_rxns=False, penalties=None, **solver_parameters)[source]¶ runs the SMILEY algorithm to determine which gaps should be filled in order for the model to create the metabolite with the given metabolite_id.
This function is good for running the algorithm once. For more fine- grained control, create a SUXModelMILP object, add a demand reaction for the given metabolite_id, and call the solve function on the SUXModelMILP object.
-
class
cobra.flux_analysis.gapfilling.SUXModelMILP(model, Universal=None, threshold=0.05, penalties=None, dm_rxns=True, ex_rxns=False)[source]¶ Bases:
cobra.core.Model.ModelModel with additional Universal and Exchange reactions. Adds corresponding dummy reactions and dummy metabolites for each added reaction which are used to impose MILP constraints to minimize the total number of added reactions. See the figure for more information on the structure of the matrix.
cobra.flux_analysis.loopless module¶
-
cobra.flux_analysis.loopless.construct_loopless_model(cobra_model)[source]¶ construct a loopless model
This adds MILP constraints to prevent flux from proceeding in a loop, as done in http://dx.doi.org/10.1016/j.bpj.2010.12.3707 Please see the documentation for an explanation of the algorithm.
This must be solved with an MILP capable solver.
cobra.flux_analysis.moma module¶
-
cobra.flux_analysis.moma.create_euclidian_distance_objective(n_moma_reactions)[source]¶ returns a matrix which will minimze the euclidian distance
This matrix has the structure [ I -I] [-I I] where I is the identity matrix the same size as the number of reactions in the original model.
- n_moma_reactions: int
- This is the number of reactions in the MOMA model, which should be twice the number of reactions in the original model
-
cobra.flux_analysis.moma.create_euclidian_moma_model(cobra_model, wt_model=None, **solver_args)[source]¶
cobra.flux_analysis.parsimonious module¶
-
cobra.flux_analysis.parsimonious.optimize_minimal_flux(cobra_model, already_irreversible=False, fraction_of_optimum=1.0, solver=None, desired_objective_value=None, **optimize_kwargs)[source]¶ Perform basic pFBA (parsimonius FBA) and minimize total flux.
The function attempts to act as a drop-in replacement for optimize. It will make the reaction reversible and perform an optimization, then force the objective value to remain the same and minimize the total flux. Finally, it will convert the reaction back to the irreversible form it was in before. See http://dx.doi.org/10.1038/msb.2010.47
cobra_model :
Modelobject- already_irreversible : bool, optional
- By default, the model is converted to an irreversible one. However, if the model is already irreversible, this step can be skipped
- fraction_of_optimum : float, optional
- Fraction of optimum which must be maintained. The original objective reaction is constrained to be greater than maximal_value * fraction_of_optimum. By default, this option is specified to be 1.0
- desired_objective_value : float, optional
- A desired objective value for the minimal solution that bypasses the initial optimization result.
- solver : string of solver name
- If None is given, the default solver will be used.
Updates everything in-place, returns model to original state at end.
cobra.flux_analysis.phenotype_phase_plane module¶
-
cobra.flux_analysis.phenotype_phase_plane.calculate_phenotype_phase_plane(model, reaction1_name, reaction2_name, reaction1_range_max=20, reaction2_range_max=20, reaction1_npoints=50, reaction2_npoints=50, solver=None, n_processes=1, tolerance=1e-06)[source]¶ calculates the growth rates while varying the uptake rates for two reactions.
Returns: a phenotypePhasePlaneData object containing the growth rates for the uptake rates. To plot the result, call the plot function of the returned object.
Example: >>> import cobra.test >>> model = cobra.test.create_test_model("textbook") >>> ppp = calculate_phenotype_phase_plane(model, "EX_glc__D_e", "EX_o2_e") >>> ppp.plot()
-
class
cobra.flux_analysis.phenotype_phase_plane.phenotypePhasePlaneData(reaction1_name, reaction2_name, reaction1_range_max, reaction2_range_max, reaction1_npoints, reaction2_npoints)[source]¶ Bases:
objectclass to hold results of a phenotype phase plane analysis
-
plot_matplotlib(theme='Paired', scale_grid=False)[source]¶ Use matplotlib to plot a phenotype phase plane in 3D.
theme: color theme to use (requires palettable)
returns: maptlotlib 3d subplot object
-
cobra.flux_analysis.reaction module¶
-
cobra.flux_analysis.reaction.assess(model, reaction, flux_coefficient_cutoff=0.001, solver=None)[source]¶ Assesses the capacity of the model to produce the precursors for the reaction and absorb the production of the reaction while the reaction is operating at, or above, the specified cutoff.
model: A
Modelobjectreaction: A
Reactionobjectflux_coefficient_cutoff: Float. The minimum flux that reaction must carry to be considered active.
solver : String or solver name. If None, the default solver will be used.
returns: True if the model can produce the precursors and absorb the products for the reaction operating at, or above, flux_coefficient_cutoff. Otherwise, a dictionary of {‘precursor’: Status, ‘product’: Status}. Where Status is the results from assess_precursors and assess_products, respectively.
-
cobra.flux_analysis.reaction.assess_precursors(model, reaction, flux_coefficient_cutoff=0.001, solver=None)[source]¶ Assesses the ability of the model to provide sufficient precursors for a reaction operating at, or beyond, the specified cutoff.
model: A
Modelobjectreaction: A
Reactionobjectflux_coefficient_cutoff: Float. The minimum flux that reaction must carry to be considered active.
solver : String or solver name. If None, the default solver will be used.
returns: True if the precursors can be simultaneously produced at the specified cutoff. False, if the model has the capacity to produce each individual precursor at the specified threshold but not all precursors at the required level simultaneously. Otherwise a dictionary of the required and the produced fluxes for each reactant that is not produced in sufficient quantities.
-
cobra.flux_analysis.reaction.assess_products(model, reaction, flux_coefficient_cutoff=0.001, solver=None)[source]¶ Assesses whether the model has the capacity to absorb the products of a reaction at a given flux rate. Useful for identifying which components might be blocking a reaction from achieving a specific flux rate.
model: A
Modelobjectreaction: A
Reactionobjectflux_coefficient_cutoff: Float. The minimum flux that reaction must carry to be considered active.
solver : String or solver name. If None, the default solver will be used.
returns: True if the model has the capacity to absorb all the reaction products being simultaneously given the specified cutoff. False, if the model has the capacity to absorb each individual product but not all products at the required level simultaneously. Otherwise a dictionary of the required and the capacity fluxes for each product that is not absorbed in sufficient quantities.
cobra.flux_analysis.single_deletion module¶
-
cobra.flux_analysis.single_deletion.single_deletion(cobra_model, element_list=None, element_type='gene', **kwargs)[source]¶ Wrapper for single_gene_deletion and single_reaction_deletion
Deprecated since version 0.4: Use single_reaction_deletion and single_gene_deletion
-
cobra.flux_analysis.single_deletion.single_gene_deletion(cobra_model, gene_list=None, solver=None, method='fba', **solver_args)[source]¶ sequentially knocks out each gene in a model
gene_list: list of gene_ids or cobra.Gene
method: “fba” or “moma”
returns ({gene_id: growth_rate}, {gene_id: status})
-
cobra.flux_analysis.single_deletion.single_gene_deletion_fba(cobra_model, gene_list, solver=None, **solver_args)[source]¶
-
cobra.flux_analysis.single_deletion.single_gene_deletion_moma(cobra_model, gene_list, solver=None, **solver_args)[source]¶
-
cobra.flux_analysis.single_deletion.single_reaction_deletion(cobra_model, reaction_list=None, solver=None, method='fba', **solver_args)[source]¶ sequentially knocks out each reaction in a model
reaction_list: list of reaction_ids or cobra.Reaction
method: “fba” or “moma”
returns ({reaction_id: growth_rate}, {reaction_id: status})
-
cobra.flux_analysis.single_deletion.single_reaction_deletion_fba(cobra_model, reaction_list, solver=None, **solver_args)[source]¶ sequentially knocks out each reaction in a model using FBA
reaction_list: list of reaction_ids or cobra.Reaction
method: “fba” or “moma”
returns ({reaction_id: growth_rate}, {reaction_id: status})
-
cobra.flux_analysis.single_deletion.single_reaction_deletion_moma(cobra_model, reaction_list, solver=None, **solver_args)[source]¶ sequentially knocks out each reaction in a model using MOMA
reaction_list: list of reaction_ids or cobra.Reaction
returns ({reaction_id: growth_rate}, {reaction_id: status})
cobra.flux_analysis.summary module¶
cobra.flux_analysis.variability module¶
-
cobra.flux_analysis.variability.calculate_lp_variability(lp, solver, cobra_model, reaction_list, **solver_args)[source]¶ calculate max and min of selected variables in an LP
-
cobra.flux_analysis.variability.find_blocked_reactions(cobra_model, reaction_list=None, solver=None, zero_cutoff=1e-09, open_exchanges=False, **solver_args)[source]¶ Finds reactions that cannot carry a flux with the current exchange reaction settings for cobra_model, using flux variability analysis.
-
cobra.flux_analysis.variability.flux_variability_analysis(cobra_model, reaction_list=None, fraction_of_optimum=1.0, solver=None, objective_sense='maximize', **solver_args)[source]¶ Runs flux variability analysis to find max/min flux values
cobra_model :
Model:- reaction_list : list of
Reaction: or their id’s - The id’s for which FVA should be run. If this is None, the bounds will be comptued for all reactions in the model.
- fraction_of_optimum : fraction of optimum which must be maintained.
- The original objective reaction is constrained to be greater than maximal_value * fraction_of_optimum
- solver : string of solver name
- If None is given, the default solver will be used.
- reaction_list : list of
Module contents¶
cobra.io package¶
Submodules¶
cobra.io.json module¶
-
cobra.io.json.load_json_model(file_name)[source]¶ Load a cobra model stored as a json file
file_name : str or file-like object
cobra.io.mat module¶
-
cobra.io.mat.from_mat_struct(mat_struct, model_id=None)[source]¶ create a model from the COBRA toolbox struct
The struct will be a dict read in by scipy.io.loadmat
-
cobra.io.mat.load_matlab_model(infile_path, variable_name=None)[source]¶ Load a cobra model stored as a .mat file
infile_path : str
- variable_name : str, optional
- The variable name of the model in the .mat file. If this is not specified, then the first MATLAB variable which looks like a COBRA model will be used
-
cobra.io.mat.model_to_pymatbridge(model, variable_name='model', matlab=None)[source]¶ send the model to a MATLAB workspace through pymatbridge
This model can then be manipulated through the COBRA toolbox
- variable_name: str
- The variable name to which the model will be assigned in the MATLAB workspace
- matlab: None or pymatbridge.Matlab instance
- The MATLAB workspace to which the variable will be sent. If this is None, then this will be sent to the same environment used in IPython magics.
cobra.io.sbml module¶
-
cobra.io.sbml.add_sbml_species(sbml_model, cobra_metabolite, note_start_tag, note_end_tag, boundary_metabolite=False)[source]¶ A helper function for adding cobra metabolites to an sbml model.
sbml_model: sbml_model object
cobra_metabolite: a cobra.Metabolite object
note_start_tag: the start tag for parsing cobra notes. this will eventually be supplanted when COBRA is worked into sbml.
note_end_tag: the end tag for parsing cobra notes. this will eventually be supplanted when COBRA is worked into sbml.
-
cobra.io.sbml.create_cobra_model_from_sbml_file(sbml_filename, old_sbml=False, legacy_metabolite=False, print_time=False, use_hyphens=False)[source]¶ convert an SBML XML file into a cobra.Model object. Supports SBML Level 2 Versions 1 and 4. The function will detect if the SBML fbc package is used in the file and run the converter if the fbc package is used.
sbml_filename: String.
old_sbml: Boolean. Set to True if the XML file has metabolite formula appended to metabolite names. This was a poorly designed artifact that persists in some models.
legacy_metabolite: Boolean. If True then assume that the metabolite id has the compartment id appended after an underscore (e.g. _c for cytosol). This has not been implemented but will be soon.
print_time: deprecated
use_hyphens: Boolean. If True, double underscores (__) in an SBML ID will be converted to hyphens
-
cobra.io.sbml.get_libsbml_document(cobra_model, sbml_level=2, sbml_version=1, print_time=False, use_fbc_package=True)[source]¶ Return a libsbml document object for writing to a file. This function is used by write_cobra_model_to_sbml_file().
-
cobra.io.sbml.parse_legacy_id(the_id, the_compartment=None, the_type='metabolite', use_hyphens=False)[source]¶ Deals with a bunch of problems due to bigg.ucsd.edu not following SBML standards
the_id: String.
the_compartment: String.
the_type: String. Currently only ‘metabolite’ is supported
use_hyphens: Boolean. If True, double underscores (__) in an SBML ID will be converted to hyphens
-
cobra.io.sbml.parse_legacy_sbml_notes(note_string, note_delimiter=':')[source]¶ Deal with legacy SBML format issues arising from the COBRA Toolbox for MATLAB and BiGG.ucsd.edu developers.
-
cobra.io.sbml.read_legacy_sbml(filename, use_hyphens=False)[source]¶ read in an sbml file and fix the sbml id’s
-
cobra.io.sbml.write_cobra_model_to_sbml_file(cobra_model, sbml_filename, sbml_level=2, sbml_version=1, print_time=False, use_fbc_package=True)[source]¶ Write a cobra.Model object to an SBML XML file.
cobra_model:
Modelobjectsbml_filename: The file to write the SBML XML to.
sbml_level: 2 is the only level supported at the moment.
sbml_version: 1 is the only version supported at the moment.
- use_fbc_package: Boolean.
- Convert the model to the FBC package format to improve portability. http://sbml.org/Documents/Specifications/SBML_Level_3/Packages/Flux_Balance_Constraints_(flux)
TODO: Update the NOTES to match the SBML standard and provide support for Level 2 Version 4
cobra.io.sbml3 module¶
-
cobra.io.sbml3.clip(string, prefix)[source]¶ clips a prefix from the beginning of a string if it exists
>>> clip("R_pgi", "R_") "pgi"
Module contents¶
cobra.manipulation package¶
Submodules¶
cobra.manipulation.annotate module¶
-
cobra.manipulation.annotate.add_SBO(model)[source]¶ adds SBO terms for demands and exchanges
This works for models which follow the standard convention for constructing and naming these reactions.
The reaction should only contain the single metabolite being exchanged, and the id should be EX_metid or DM_metid
cobra.manipulation.delete module¶
-
cobra.manipulation.delete.delete_model_genes(cobra_model, gene_list, cumulative_deletions=True, disable_orphans=False)[source]¶ delete_model_genes will set the upper and lower bounds for reactions catalysed by the genes in gene_list if deleting the genes means that the reaction cannot proceed according to cobra_model.reactions[:].gene_reaction_rule
cumulative_deletions: False or True. If True then any previous deletions will be maintained in the model.
-
cobra.manipulation.delete.find_gene_knockout_reactions(cobra_model, gene_list, compiled_gene_reaction_rules=None)[source]¶ identify reactions which will be disabled when the genes are knocked out
cobra_model:
Modelgene_list: iterable of
Gene- compiled_gene_reaction_rules: dict of {reaction_id: compiled_string}
- If provided, this gives pre-compiled gene_reaction_rule strings. The compiled rule strings can be evaluated much faster. If a rule is not provided, the regular expression evaluation will be used. Because not all gene_reaction_rule strings can be evaluated, this dict must exclude any rules which can not be used with eval.
-
cobra.manipulation.delete.get_compiled_gene_reaction_rules(cobra_model)[source]¶ Generates a dict of compiled gene_reaction_rules
Any gene_reaction_rule expressions which cannot be compiled or do not evaluate after compiling will be excluded. The result can be used in the find_gene_knockout_reactions function to speed up evaluation of these rules.
-
cobra.manipulation.delete.prune_unused_metabolites(cobra_model)[source]¶ Removes metabolites that aren’t involved in any reactions in the model
cobra_model: A Model object.
-
cobra.manipulation.delete.prune_unused_reactions(cobra_model)[source]¶ Removes reactions from cobra_model.
cobra_model: A Model object.
reactions_to_prune: None, a string matching a reaction.id, a cobra.Reaction, or as list of the ids / Reactions to remove from cobra_model. If None then the function will delete reactions that have no active metabolites in the model.
cobra.manipulation.modify module¶
-
cobra.manipulation.modify.canonical_form(model, objective_sense='maximize', already_irreversible=False, copy=True)[source]¶ Return a model (problem in canonical_form).
Converts a minimization problem to a maximization, makes all variables positive by making reactions irreversible, and converts all constraints to <= constraints.
model: class:~cobra.core.Model. The model/problem to convert.
objective_sense: str. The objective sense of the starting problem, either ‘maximize’ or ‘minimize’. A minimization problems will be converted to a maximization.
already_irreversible: bool. If the model is already irreversible, then pass True.
copy: bool. Copy the model before making any modifications.
-
cobra.manipulation.modify.convert_to_irreversible(cobra_model)[source]¶ Split reversible reactions into two irreversible reactions
These two reactions will proceed in opposite directions. This guarentees that all reactions in the model will only allow positive flux values, which is useful for some modeling problems.
cobra_model: A Model object which will be modified in place.
-
cobra.manipulation.modify.initialize_growth_medium(cobra_model, the_medium='MgM', external_boundary_compartment='e', external_boundary_reactions=None, reaction_lower_bound=0.0, reaction_upper_bound=1000.0, irreversible=False, reactions_to_disable=None)[source]¶ Sets all of the input fluxes to the model to zero and then will initialize the input fluxes to the values specified in the_medium if it is a dict or will see if the model has a composition dict and use that to do the initialization.
cobra_model: A cobra.Model object.
the_medium: A string, or a dictionary. If a string then the initialize_growth_medium function expects that the_model has an attribute dictionary called media_compositions, which is a dictionary of dictionaries for various medium compositions. Where a medium composition is a dictionary of external boundary reaction ids for the medium components and the external boundary fluxes for each medium component.
external_boundary_compartment: None or a string. If not None then it specifies the compartment in which to disable all of the external systems boundaries.
external_boundary_reactions: None or a list of external_boundaries that are to have their bounds reset. This acts in conjunction with external_boundary_compartment.
reaction_lower_bound: Float. The default value to use for the lower bound for the boundary reactions.
reaction_upper_bound: Float. The default value to use for the upper bound for the boundary.
irreversible: Boolean. If the model is irreversible then the medium composition is taken as the upper bound
reactions_to_disable: List of reactions for which the upper and lower bounds are disabled. This is superceded by the contents of media_composition
cobra.manipulation.validate module¶
Module contents¶
cobra.topology package¶
Submodules¶
cobra.topology.reporter_metabolites module¶
-
cobra.topology.reporter_metabolites.identify_reporter_metabolites(cobra_model, reaction_scores_dict, number_of_randomizations=1000, scoring_metric='default', score_type='p', entire_network=False, background_correction=True, ignore_external_boundary_reactions=False)[source]¶ Calculate the aggregate Z-score for the metabolites in the model. Ignore reactions that are solely spontaneous or orphan. Allow the scores to have multiple columns / experiments. This will change the way the output is represented.
cobra_model: A cobra.Model object
TODO: CHANGE TO USING DICTIONARIES for the_reactions: the_scores
reaction_scores_dict: A dictionary where the keys are reactions in cobra_model.reactions and the values are the scores. Currently, only supports a single numeric value as the value; however, this will be updated to allow for lists
number_of_randomizations: Integer. Number of random shuffles of the scores to assess which are significant.
scoring_metric: default means divide by k**0.5
score_type: ‘p’ Is the only option at the moment and indicates p-value.
entire_network: Boolean. Currently, only compares scores calculated from the_reactions
background_correction: Boolean. If True apply background correction to the aggreagate Z-score
ignore_external_boundary_reactions: Not yet implemented. Boolean. If True do not count exchange reactions when calculating the score.