Welcome to miRPursuit | sRNA-workflow’s documentation!¶
Attention
sRNA-workflow is now called miRPursuit.
Important
This is still under construction. But we are working hard to complete this page.
Brief description¶
This is an open source project, developed at the Forest Biotech lab group ITQB-NOVA. miRPursuit is a pipeline suited for running end-to-end analysis of small RNA (sRNA) data, from the raw data to the annotated reads. You can execute this pipeline in parallel for all your libraries, ensuring that all libraries are treated in the same way, thus providing a high degree of reproducibility in your analysis. The execution of this program produces a detailed documentation of all the parameters used, visible in the logging files. Another major feature is that all the steps can be customized, making it particularly useful when working with non-model organisms.
Guide¶
Introduction¶
miRPursuit is an open source project developed by Forest Biotech group to automate the management of workloads in
Objectives¶
The main goal of this pipeline is to aggregate independent modules into a pipeline to automate the process of extracting information from sequence data.
Stages¶
This is an overview of the organizational structure of miRPursuit. This is useful if you want to re-do the analysis of the pipeline with different parameters only from a specific stage onward. This way you can avoid unnecessary repeating of stages.
- The workflow is divided in 4 main stages:

Image 1 - miRPursuit general schema.
Pre-preprocessing¶
There are multiple entry points depending on the form of the raw data. Some NGS sequencing service providers might ship your data already trimmed for adaptors, or you might want to use the raw data provided directly by the sequencing equipment, or you might want to use fasta files compiled from another source.
By using miRPursuit you can specify the type of input file you will use. The most simple is the - -fasta flag that searches the inserts_dir path ( see config files workdir.cfg ) for the target .fa/.fasta libraries and makes a copy to the project folder. In case no .fa/.fasta files are found the program will also search for compressed .fa.gz/.fasta.gz files and proceed to uncompress them.
In case the libraries are still in the fastq format the - -fastq flag should be given. This method does a quality control (fastqc not yet but soon) and then converts the fastq libraries to .fasta, analogously to what is done with “fasta” files, compressed fq.gz/fastq.gz files will be uncompressed if no .fq/.fastq file is found.
Additionally, the - -trim flag can be set to remove adaptor sequences. This requires the adaptor sequence to be stored in the adaptor var (see config files workdir.cfg ).
Filtering¶
- Filtering Databases
- The fasta sequences are filtered based on their length, abundance, low complexity and t/r RNA are removed. These parameters can be set in the wbench_filter.cfg configuration file.
- Genome and miRBase
- The reads are further filtered by mapping them to the setup genome file with ‘0’ mismatches using patman. These parameters can be set in the patman_genome.cfg configuration file.
Annotation¶
- Identification of conserved miRNAs (miRBase)
- The mapped reads are then aligned to the miRBase⁺ (ref) database using miRProf with the parameter set in the wbench_mirprof.cfg configuration file. The genome mapped reads are separated into two files per library those that mapped with miRBase (conserved reads) and those that did not (non conserved reads).
- tasiRNA prediction
- The non conserved reads are run through the ta-si predictor to identify trans acting siRNA (tasi-RNA) using the parameters in the wbench_tasi.cfg
- Novel miRNA
- The non conserved reads are also used to predict novel miRNA with miRCat by searching the genome for their respective precursor nucleotide sequences in the setup genome file. The parameters used by miRCat are set in the wbench_mircat.cfg configuration file and the genome file is set in the workdirs.cfg . If memory (RAM) restrictions apply, the genome can be split into several parts and miRCat will be run once for each part. The various parts should all be held in the same directory with a common name which includes the word part and the sequential number. Afterwards the resulting files will be merged and filtered to remove miRNAs that paired with more genome sites than those specified in the configuration file wbench_mircat.cfg.
Reporting¶
- Merging results and stepwise stats
- The number of sequences kept in each step are given for each library, both total numbers and distinct numbers of sequences. The identified sequences and their respective absolute count are stored in a tab separate value file (.tsv). This provides easy exportation to most statistical softwares as well as MS Excel.
Targets¶
- Validation of targets
- Target validation is done based on the supplied degradome and transcriptome information, which are both necessary to perform this analysis. The file paths are stored in the workdirs.cfg configuration file and the parameters are stored in wbench_paresnip.cfg configuration file.
Installation¶
This pipeline is intended to be run in a Linux environment. Installation can be accomplished by grabbing a copy of the project from github and then running the installation script. Below are a few lines to help guide you through the installation process.
- The pipeline is optimized to run in the command line as detailed below.
- To open a terminal on a debian based system press: CTRL + ALT + T
- To install on a remote machine, make a connection via ssh and navigate to the desired path of installation.
Download program¶
Grab a copy from `github <https://github.com/forestbiotech-lab/miRPrusuit>`_
- If you have git installed on you machine.
Simply navigate to your chosen directory:
#Clone project with git #This might prompt you for your github credentials. If authentication fails #try again and it should run without any prompts. git clone https://github.com/forestbiotech-lab/miRPrusuit cd miRPursuit
- Without git installed on your system. Installing using git clone is preferred since it allows updating to remove bugs or get new features.
Simply visit github project and download .zip file.
#Download through command line wget https://github.com/forestbiotech-lab/miRPursuit/archive/master.zip -O miRPursuit.zip
Extract contents to chosen directory
#Extract contents of archive unzip miRPursuit.zip
Install Script¶
Run the install.sh file:
cd [toPath]/miRPrusuit
./install.sh
Important
Make sure you restart your terminal/computer to update your path so PatMaN can be accessed.
Alternatively you can source your startup shell file. Example for Bash shell.:
source ~/.bashrc
Congratulations you should now have miRPursuit installed in your system.
Check help section for information on help and how to send feedback about this project.
- Configure parameters
Now check that all the parameters are set to your convenience.
Go to configuration page.
Dependencies¶
Path¶
To ensure persistence of environmental variables (PATH) throughout sessions, the paths to the dependencies are stored in shell scripts. The installation script checks which shell is being used by the system and saves the path to the corresponding initiation shell script.
Table 1 shows which shells are contemplated in the installation script. If your system uses a shell that is not listed (others) then the file $HOME/.profile is created. You should ensure that your shell is reading given shell script. In case your shell is not using the listed shell file, add the following line of code, to a shell script that is executed on startup of the shell you use.:
source $HOME/.profile
Table 1 - List of shells and it’s associated shell script.
| Shell | Shell scripts |
|---|---|
| bash | $HOME/.bashrc |
| zsh | $HOME/.zshrc |
| fish | $HOME/.config/fish/config.fish |
| ksh | $HOME/.profile |
| tcsh | $HOME/.login |
| others | $HOME/.profile |
Detailed installation guide¶
Step by step guide through installation script.
Installation of dependencies¶
The default directory for storing dependencies is ${HOME}/.Software, it will be created if it doesn’t exist. To use another directory change the variable SOFTWARE in software_dirs.cfg.
The installation script starts by checking if PatMaN is installed on the system. If it is not available on the system it will be downloaded to the directory in the variable SOFTWARE. The downloaded archive is extracted and added to the path.
miRPursuit works best with Oracle’s Java v.8. So instead of changing your system’s installed Java VM miRPursuit uses the Java VM in the variable JAVA_DIR in software_dirs.cfg. If the variable is empty the installation script will download Java, extract it and set JAVA_DIR variable to the correct directory.
If fastq_to_fasta from FASTX-Toolkit is not on available on the system it will be downloaded to the directory in the variable SOFTWARE. The downloaded archive will extracted and added to the path.
UEA sRNA workbench is run by miRPursuit from the WBENCH_DIR variable in software_dirs.cfg. If the variable isn’t set the installation script will download the workbench and set up the variable. Since usage of UEA sRNA workbench requires acceptance of it’s terms of use. On your first run you will be prompted to read and accept their term of use. Alternatively you can run their GUI and accept their terms of use in a graphical environment.
Setting variables in workdirs.cfg¶
This section will guide you through the command prompts that will be issued.
Create source data folder? This creates a directory for storing resources such as genomes, miRBase, etc. As a good practise it is recommend to store every thing in a common folder structure. Default is $HOME/source_data
- Y|y - Default directory is created.
- N|n - Specify an alternate directory.
Config files¶
There are three types of config files, General use, Module specific and System parameters.
- General use
- Are those that are used by the main script to feed specific locations or general configuration parameters.
- Module specific
- Are configurations that are used by the module. The names of these config files start with the wbench prefix.
- System parameters
- These configs hold the values of colors and others miscellaneous variables for ease of access.
General use¶
These two config files should be properly configured, to ensure the program runs. The install script will fill out all variables.
software_dirs.cfg:
#Path were install script will install software SOFTWARE= #Path to workbench http://srna-workbench.cmp.uea.ac.uk/ WBENCH_DIR= #Path to java use 1.7 or greater JAVA_DIR= #Number of times program has been run RUN=0The SOFTWARE variable is the path to the directory were the install script will install all necessary dependancies. WBENCH_DIR
workdirs.cfg:
#LINES THE SWITH # ARE INFORMATIONAL ONLY #Workdir is the path to the directory where this program will run data #workdir must end with trailing "/" workdir=${HOME}/miRPursuit_Projects/miRtest/ #Path to the mirbase database. Go to http://www.mirbase.org or download latest from: ftp://mirbase.org/pub/mirbase/CURRENT/ MIRBASE=${source_data}/mirbase/mature.fa #Used by java MEMORY="4g" #Set this to the max number of processed that can be used THREADS=2 #Path to the directory where input data is located #Test directory in [pathToMiRPursuit]/testDataset INSERTS_DIR=${SOURCE_DATA}/sRNA/ #Path to the genome to be used #Test genome can be found here [pathToMiRPursuit]/testDataset/Genome/Arabidopsis_thaliana.TAIR10.dna_rm.chromosome.4.fa GENOME=${SOURCE_DATA}/genomes/my_genome.fa #Path to the genome to be used by mircat. Leave this, as ${GENOME} if no memory resctrictions apply to your case. Check manual on using parts GENOME_MIRCAT=${GENOME/.fa/part-1.fa} #The suffix of the filter to be used. Check /config/workbench_filter_*.cfg FILTER_SUF=18_26_5 #Adaptor trimming #You must set the --trim flag ADAPTOR="TGGAATTCTCGGGTGCCAAGG" #Deprecated - Soon removed LCSCIENCE_LIB= #These var are only used for target prediction (PAREsnip) TRANSCRIPTOME= DEGRADOME=
Module specific¶
There is a config file for each module in the miRPursuit/config directory. The default values are posted, for further reference, please consult the website of the respective tool.
wbench_filter.cfg - Filter your sRNA sequences. Length, abundance, T/R RNA:
#Broad range default values min_length=18 max_length=26 min_abundance=5 max_abundance=2147483647 norm_abundance=false filter_low_comp=true filter_invalid=true trrna=true trrna_sense_only=false filter_genome_hits=false filter_norm_abund=false filter_kill_list=false add_discard_log=false genome=null kill_list=null discard_log=nullwbench_mircat.cfg - miRCat predict novel miRNAs through alignment with genome to find putative precursors:
#Default values (Broad) extend=100.0 min_energy=-25.0 min_paired=17 max_gaps=3 max_genome_hits=16 min_length=18 max_length=26 min_gc=20 max_unpaired=60 max_overlap_percentage=80 min_locus_size=1 orientation=80 min_hairpin_len=60 complex_loops=true pval=0.05 min_abundance=1 cluster_sentinel=200 Thread_Count=12 #Default (plants) extend=100.0 min_energy=-25.0 min_paired=17 max_gaps=3 max_genome_hits=16 min_length=20 max_length=22 min_gc=20 max_unpaired=50 max_overlap_percentage=80 min_locus_size=1 orientation=80 min_hairpin_len=60 complex_loops=true pval=0.05 min_abundance=1 cluster_sentinel=200 Thread_Count=20wbench_mirprof.cfg - miRProf identifies conserved miRNA, through alignment to the miRBase database of miRNA:
#Default values mismatches=0 overhangs=true group_mismatches=true group_organisms=true group_variant=true group_mature_and_star=false only_keep_best=true min_length=18 max_length=26 min_abundance=5wbench_tasi.cfg - ta-si predictor, identifies phased 21nt sRNAs characteristic of ta-siRNA loci:
#Default values p_val_threshold=1.0E-4 min_abundance=2paresnip.cfg - PAREsnip validates targets of regulation by sRNAs requires degradome and a transcriptome sequences:
#Default values min_sRNA_abundance=5 subsequences_are_secondary_hits=false output_secondary_hits_to_file=false use_weighted_fragments_abundance=true category_0=true category_1=true category_2=true category_3=true category_4=false discard_tr_rna=true discard_low_complexity_srnas=false discard_low_complexity_candidates=false min_fragment_length=20 max_fragment_length=21 min_sRNA_length=19 max_sRNA_length=24 allow_single_nt_gap=false allow_mismatch_position_11=false allow_adjacent_mismatches=false max_mismatches=4.0 calculate_pvalues=true number_of_shuffles=100 pvalue_cutoff=0.05 do_not_include_if_greater_than_cutoff=true number_of_threads=23 auto_output_tplot_pdf=falsepatman_genome.cfg - Patman a pattern matcher for short sequences:
#Default values #Set maximum edit distance to N (Default: 0) EDITS=0 #Set maximum number of gaps to N (default: 0) GAPS=0 #Do not match reverse-complements (default: FALSE) SINGLESTRAND=FALSE #Prefetch N nodes (default: 3) Related with performance PREFETCH=3 ################# #Not implemented# ################# #Interpret ambiguity codes in patterns (Flag for using ambicodes) #ambicodes=FALSE
System parameters¶
These are generally hardcoded, don’t change these unless you know what you are doing.
- term-colors.cfg - Colors for terminal and other useful vars.
Getting Started¶
So at this point you have already completely installed <install.html>_ miRPursuit. You should be familiar with the various stages <stages.html>_ in the pipeline and have step up all the necessary configuration files. You are now ready to run miRPursuit for the first time.
You can run with these default settings or you can start with your customized settings. MiRBase will be downloaded by the install script but you should have a genome file or download one to run (Try Phytozome or ensemble plants). For the purpose of this example all instructions will be based on the following configurations in Table 3.
Table 3 - Example configuration of workdirs.cfg
| Variable | Value |
|---|---|
| workdir | ${HOME}/miRtest/ |
| MEMORY | “4g” |
| THREADS | 2 |
| INSERTS_DIR | ${miRPursuit}/testDataset/ |
| GENOME | ${SOURCE_DATA}/Genome/Genome.fa |
| GENOME_MIRCAT | ${GENOME} |
| FILTER_SUF | 18_26_5 |
| ADAPTOR | “TGGAATTCTCGGGTGCCAAGG” |
Bash variables in table 3 and their values:
(Keep it simple, store all dependant DBs in $SOURCE_DATA it will be simpler to configure. But substitute appropriately.)
SOURCE_DATA=${HOME}/souce_data
(This is the path used in this example. Depending on where you stored your installation, you should substitute appropriately.)
miRPursuit=${HOME}/git/miRPursuit
(In this case we don't specify a user. In reality this would expand to your home dir. /home/[user])
HOME=/home/
How to run the program¶
Run the command:
${miRPursuit}/./miRPursuit.sh -f 1 -l 2 --fasta test_dataset-
- This is the simplest test case. Let’s break down this command.
- ${miRPursuit}/./miRPursuit.sh - This is used to execute the main script to start miRPursuit. You can simply run ./miRPursuit if you’re current path is already in the miRPursuit directory.
- -f - The number of the first library
- -l - The number of the last library
- –fasta test_dataset- - Run in fasta mode, and use all libraries that have the string “test_dataset-” preceding the sequential numbering.
Important
MiRPursuit is designed to run an interval of libraries. So it will run all libraries starting with test_dataset-1.fa (-f first in this example 1), up to test_dataset-1.fa (-l last in this example 2). The files to be processed in your INSERTS_DIR should have a common string along with a sequential numbering.
However you can also use it in specific file mode. That is specify the file you want to process for a single run.
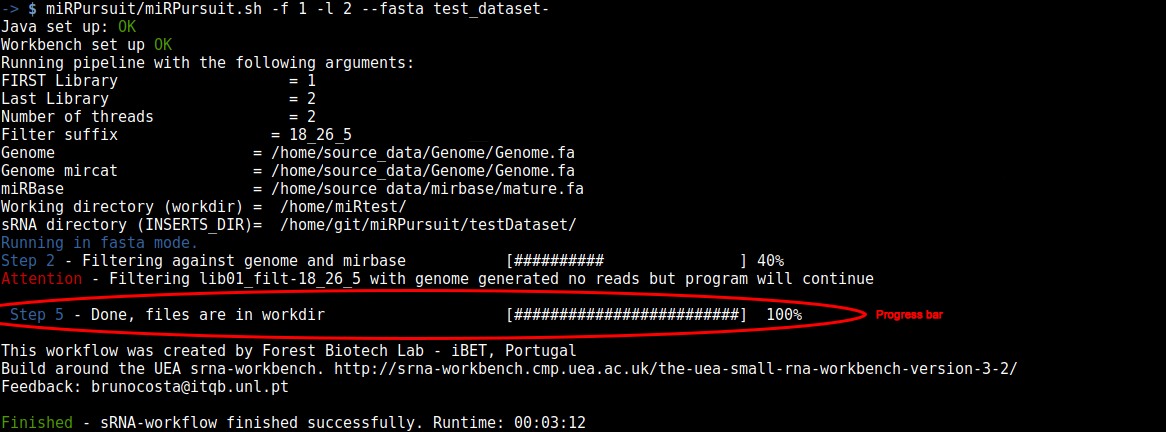
Figure 2 - miRPursuit full run in fasta mode for libraries 1 and 2.
In Figure 2, a complete run of miRPursuit is shown using the above specified command. The first procedure the pipeline does is to check that all variables are defined and their values link to existing files. If any of the mandatory files is missing a warning will be issued and you will have to start the run again (Fig. 3).
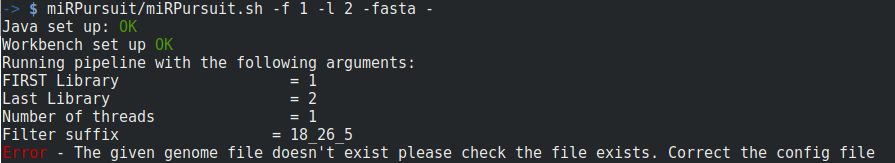
Figure 3 - Warning issued by miRPursuit due to lack of genome file.
During the miRPursuit run the progress bar will update you as to which stage is in execution, along with its percentage of conclusion. Once the run is finished a finished message will be given as shown in figure 2.
Important
Tip: In order to run on remote servers without the need to stay connected try screen
Run options¶
- The full listing of the options available
- -f|–lib-first First library to be processed.
- -l|–lib-last Last library to be processed.
- -h|–help See the list of options.
- Optional arguments
- –lib Set the library number that should be assigned to the specified file. (Only relevant if using specific files)
- –fasta Set the program to start using fasta files. As an argument supply the file name that identifies the series to be used. Ex: Lib_1.fa, Lib_2.fa, .. –> argument should be Lib_
- –fasta (In specific mode. i.e. no -f and -l) Set the program to start using fasta files. If no sequence of libraries are given then the argument can be a specific fasta file (uncompressed for now).
- –fastq Set the program to start using fastq files. As an argument supply the file name that identifies the series to be used. Ex: Lib_1.fq, Lib_2.fq, .. –> argument should be Lib_ , if no .fq file is present but instead a .fastq.gz file will additionally be extracted automatically.
- –trim Set this flag to perform adaptor triming. No argument should be given. The adaptor is in the workdirs.cfg config file in the variable ADAPTOR.
- -s|–step Step is an optional argument used to jump steps to start the analysis from a different point
- Step 1: Adaptor trimming (If flagged) & Wbench Filter
- Step 2: Filter Genome & mirbase
- Step 3: Tasi
- Step 4: Mircat
- Step 5: Reporting
Specific file mode * –fasta${NC} (In specific mode. i.e. no -f and -l) Set the program to start using fasta files. If no sequence of libraries are given then the argument can be a specific fasta file (uncompressed for now). * –fasta${NC} (In specific mode. i.e. no -f and -l) Set the program to start using fasta files. If no sequence of libraries are given then the argument can be a specific fasta file (uncompressed for now). * –lib${NC} (Optional) (In specific mode. i.e. no -f and -l) Set the library number to be attributed to the file. Should be coupled with –fasta or –fastq.
Both fasta and fastq options work in the same manner they require the preceding string to the sequential numbering that all libraries have in common. Ex:
- Lib01.fa
- Lib02.fa
- Lib03.fa
The common string is “Lib” or “ib” or “b”, the sequential numbering is 01,02,03. And .fa is the extension.
Attention
Avoid using spaces in file names. As it might generate unexpected errors.
If the –trim flag is present in the command the reads are then searched for adaptor sequences using the fastx_clipper software of the FASTX toolkit; sRNA sequences are assumed to be the string of nucleotides between the 5’ and 3’ adaptor sequences.
Additionally if the –fastq option is used. A fastqc quality report will be generated for each of the libraries.
Help¶
Help and feature request
I’m glad to help you out, all feedback is valuable. Please open an issue on the github project. This way you can see if it is something that has already been asked by others. I will respond as soon as possible. Opening issues also allows to set milestones and to include changes to the project to accommodate requests.