

Inferring a gene regulatory network (GRN) from gene expression data is a computationally expensive task, exacerbated by increasing data sizes due to advances in high-throughput gene profiling technology.
The Arboretum software library addresses this issue by providing a computational strategy that allows executing the class of GRN inference algorithms exemplified by GENIE3 [1] on hardware ranging from a single computer to a multi-node compute cluster. This class of GRN inference algorithms is defined by a series of steps, one for each target gene in the network, where the most important candidates from a set of regulators are determined from a regression model to predict a target gene’s expression profile.
Members of the above class of GRN inference algorithms are attractive from a computational point of view because they are parallelizable by nature. In arboretum, we specify the parallelizable computation as a dask graph [2], a data structure that represents the task schedule of a computation. A dask scheduler assigns the tasks in a dask graph to the available computational resources. Arboretum uses the dask distributed scheduler to spread out the computational tasks over multiple processes running on one or multiple machines.
Arboretum currently supports 2 GRN inference algorithms:
- GRNBoost2: a novel and fast GRN inference algorithm using Stochastic Gradient Boosting Machine [3] (SGBM) regression with early-stopping regularization.
- GENIE3: the classic GRN inference algorithm using Random Forest (RF) or ExtraTrees (ET) regression.
Usage Example¶
# import python modules
import pandas as pd
from arboretum.utils import load_tf_names
from arboretum.algo import grnboost2
# load the data
ex_matrix = pd.read_csv(<ex_path>, sep='\t')
tf_names = load_tf_names(<tf_path>)
# infer the gene regulatory network
network = grnboost2(expression_data=ex_matrix,
tf_names=tf_names)
network.head()
| TF | target | importance |
|---|---|---|
| G109 | G1406 | 151.648784 |
| G16 | G1440 | 136.741815 |
| G188 | G938 | 124.707570 |
| G10 | G1312 | 124.195566 |
| G48 | G1419 | 121.488200 |
Check out more examples.
References¶
| [1] | Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P (2010) Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE |
| [2] | Rocklin, M. (2015). Dask: parallel computation with blocked algorithms and task scheduling. In Proceedings of the 14th Python in Science Conference (pp. 130-136). |
| [3] | Friedman, J. H. (2002). Stochastic gradient boosting. Computational Statistics & Data Analysis, 38(4), 367-378. |
| [4] | Marbach, D., Costello, J. C., Kuffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., … & Dream5 Consortium. (2012). Wisdom of crowds for robust gene network inference. Nature methods, 9(8), 796-804. |
Installation Guide¶
Caution
Python Environment
It is highly recommended to prepare a Python environment with the Anaconda or Miniconda distribution and install Arboretum’s dependencies using the conda package manager.
- NumPy
- SciPy
- scikit-learn
- pandas
- dask
- distributed
This avoids complexities in ensuring that libraries like NumPy and SciPy link against an optimized implementation of linear algebra routines.
Install using pip¶
The arboretum package is available from PyPI (Python Package Index), a repository of software for the Python programming language.
Using pip, installing the arboretum package is straightforward:
$ pip install arboretum
Install from source¶
Installing Arboretum from source is possible using following steps:
- clone the Github repository using the git tool:
$ git clone https://github.com/tmoerman/arboretum.git
$ cd arboretum
- build Arboretum using the provided script:
$ ./pypi_build.sh
- install the freshly built Arboretum package using pip:
$ pip install dist/*
User Guide¶
Modules overview¶
Arboretum consists of multiple python modules:
arboretum.algo¶
arboretum.core¶
- Intended for advanced users.
- Contains the low-level building blocks of the Arboretum framework.
arboretum.utils¶
- Contains small utility functions.
Dependencies Overview¶
Arboretum uses well-established libraries from the Python ecosystem. Arboretum avoids being a proverbial “batteries-included” library, as such an approach often entails unnecessary complexity and maintenance. Arboretum aims at doing only one thing, and doing it well.
Concretely, the user will be exposed to one or more of following dependencies:
format. Pandas and NumPy are well equipped with functions for data preprocessing. * Dask.distributed: to run Arboretum on a cluster, the user is responsible for setting up a network of a scheduler and workers. * scikit-learn: relevant for advanced users only. Arboretum can run “DIY” inference where the user provides their own parameters for the Random Forest or Gradient Boosting regressors.
Input / Output¶
Arboretum accepts as input:
- an expression matrix (rows = observations, columns = genes)
- (optionally) a list of gene names in the expression matrix
- (optionally) a list of transcription factors (a.k.a. TFs)
Arboretum returns as output:
Tip
As data for following code snippets, you can use the data for network 1 from the DREAM5 challenge (included in the resources folder of the Github repository):
<ex_path>= net1_expression_data.tsv<tf_path>= net1_transcription_factors.tsv
Expression matrix as a Pandas DataFrame¶
The input can be specified in a number of ways. Arguably the most straightforward way is to specify the expression matrix as a Pandas DataFrame, which also contains the gene names as the column header.
.-------------. | expression | | matrix (DF) | ---. .-----------. '-------------' \ | GRNBoost2 | .------------. :--> | or | --> | regulatory | .---------------. / | GENIE3 | | links (DF) | | transcription | -' '-----------' '------------' | factors | '---------------'
In the following code snippet, we launch network inference with grnboost2 by
specifying the expression_data as a DataFrame.
import pandas as pd
from arboretum.utils import load_tf_names
from arboretum.algo import grnboost2
# ex_matrix is a DataFrame with gene names as column names
ex_matrix = pd.read_csv(<ex_path>, sep='\t')
# tf_names is read using a utility function included in Arboretum
tf_names = load_tf_names(<tf_path>)
network = grnboost2(expression_data=ex_matrix,
tf_names=tf_names)
Expression matrix as a NumPy ndarray¶
Arboretum also supports specifying the expression matrix as a Numpy ndarray (in our case: a 2-dimensional matrix). In this case, the gene names must be specified explicitly.
.-------------. | expression | | matrix (DF) | -----. '-------------' | .-----------. .-------------. | | GRNBoost2 | .------------. | gene names | -----+--> | or | --> | regulatory | '-------------' | | GENIE3 | | links (DF) | .---------------. | '-----------' '------------' | transcription | ---' | factors | '---------------'
Caution
You must specify the gene names in the same order as their corresponding columns of the NumPy matrix. Getting this right is the user’s responsibility.
import numpy as np
from arboretum.utils import load_tf_names
from arboretum.algo import grnboost2
# ex_matrix is a numpy ndarray, which has no notion of column names
ex_matrix = np.genfromtxt(<ex_path>, delimiter='\t', skip_header=1)
# we read the gene names from the first line of the file
with open(<ex_path>) as file:
gene_names = [gene.strip() for gene in file.readline().split('\t')]
# sanity check to verify the ndarray's nr of columns equals the length of the gene_names list
assert ex_matrix.shape[1] == len(gene_names)
# tf_names is read using a utility function included in Arboretum
tf_names = load_tf_names(<tf_path>)
network = grnboost2(expression_data=ex_matrix,
gene_names=gene_names, # specify the gene_names
tf_names=tf_names)
Running with a custom Dask Client¶
Arboretum uses Dask.distributed to parallelize its workloads. When the user doesn’t specify a dask distributed Client explicitly, Arboretum will create a LocalCluster and a Client pointing to it.
Alternatively, you can create and configure your own Client instance and pass it on to Arboretum. Situations where this is useful include:
- inferring multiple networks from different datasets
- inferring multiple networks using different parameters from the same dataset
- the user requires custom configuration for the LocalCluster (memory limit, nr of processes, etc.)
Following snippet illustrates running the gene regulatory network inference multiple times, with different initialization seed values. We create one Client and pass it to the different inference steps.
import pandas as pd
from arboretum.utils import load_tf_names
from arboretum.algo import grnboost2
from distributed import LocalCluster, Client
# create custom LocalCluster and Client instances
local_cluster = LocalCluster(n_workers=10,
threads_per_worker=1,
memory_limit=8e9)
custom_client = Client(local_cluster)
# load the data
ex_matrix = pd.read_csv(<ex_path>, sep='\t')
tf_names = load_tf_names(<tf_path>)
# run GRN inference multiple times
network_666 = grnboost2(expression_data=ex_matrix,
tf_names=tf_names,
client=custom_client, # specify the custom client
seed=666)
network_777 = grnboost2(expression_data=ex_matrix,
tf_names=tf_names,
client=custom_client, # specify the custom client
seed=777)
# close the Client and LocalCluster after use
client.close()
local_cluster.close()
Running with a Dask distributed scheduler¶
Arboretum was designed to run gene regulatory network inference in a distributed
setting. In distributed mode, some effort by the user or a systems administrator
is required to set up a dask.distributed scheduler and some workers.
Tip
Please refer to the Dask distributed network setup documentation for instructions on how to set up a Dask distributed cluster.
Following diagram illustrates a possible topology of a Dask distributed cluster.
.=[node_2]==============. .=[node_3]=========.
.=[node_1]======. | .--------------. | | .------------. |
| .--------. | | | Dask |<----+----------+--| 10 workers | |
| | Client |---+----+->| distributed |<----+--. | '------------' |
| '--------' | | | scheduler |<-. | \ '=================='
'===============' | '--------------' | | \
| | | \ .=[node_4]=========.
| .------------. | | \ | .------------. |
| | 10 workers |----' | '--+--| 10 workers | |
| '------------' | | '------------' |
'=======================' '=================='
node_1runs a Python script, console or a Jupyter notebook server, a Client instance is configured with the TCP address of the distributed scheduler, running onnode_2node_2runs a distributed scheduler and 10 workers pointing to the schedulernode_3runs 10 distributed workers pointing to the schedulernode_4runs 10 distributed workers pointing to the scheduler
With a small modification to the code, we can infer a regulatory network using all workers connected to the distributed scheduler. We specify a Client that is connected to the Dask distributed scheduler and pass it as an argument to the inference function.
import pandas as pd
from arboretum.utils import load_tf_names
from arboretum.algo import grnboost2
from distributed import Client
ex_matrix = pd.read_csv(<ex_path>, sep='\t')
tf_names = load_tf_names(<tf_path>)
scheduler_address = 'tcp://10.118.224.134:8786' # example address of the remote scheduler
cluster_client = Client(scheduler_address) # create a custom Client
network = grnboost2(expression_data=ex_matrix,
tf_names=tf_names,
client=cluster_client) # specify Client connected to the remote scheduler
GRN Inference Algorithms¶
Arboretum hosts multiple (currently 2, contributions welcome!) algorithms for inference of gene regulatory networks from high-throughput gene expression data, for example single-cell RNA-seq data.
GRNBoost2¶
GRNBoost2 is the flagship algorithm for gene regulatory network inference, hosted in the Arboretum framework. It was conceived as a fast alternative for GENIE3, in order to alleviate the processing time required for larger datasets (tens of thousands of observations).
GRNBoost2 adopts the GRN inference strategy exemplified by GENIE3, where for each gene in the dataset, the most important feature are a selected from a trained regression model and emitted as candidate regulators for the target gene. All putative regulatory links are compiled into one dataset, representing the inferred regulatory network.
In GENIE3, Random Forest regression models are trained.
DREAM5 benchmark¶
Concept and Background¶
Arboretum was conceived to address the need for a faster alternative for the classic GENIE3 implementation for inferring gene regulatory networks from high-throughput gene expression profiles.
In summary, GENIE3 performs a number of independent learning tasks. This inference “architecture” suggests two approaches for speeding up the algorithm:
- Speeding up the individual learning tasks.
- Specifying the task coordination logic so that the tasks can be executed in parallel on distributed hardware.
Page contents
FAQ¶
Q: How can I use the Dask diagnostics (bokeh) dashboard?¶
Dask distributed features a nice web interface for monitoring the execution of a Dask computation graph.
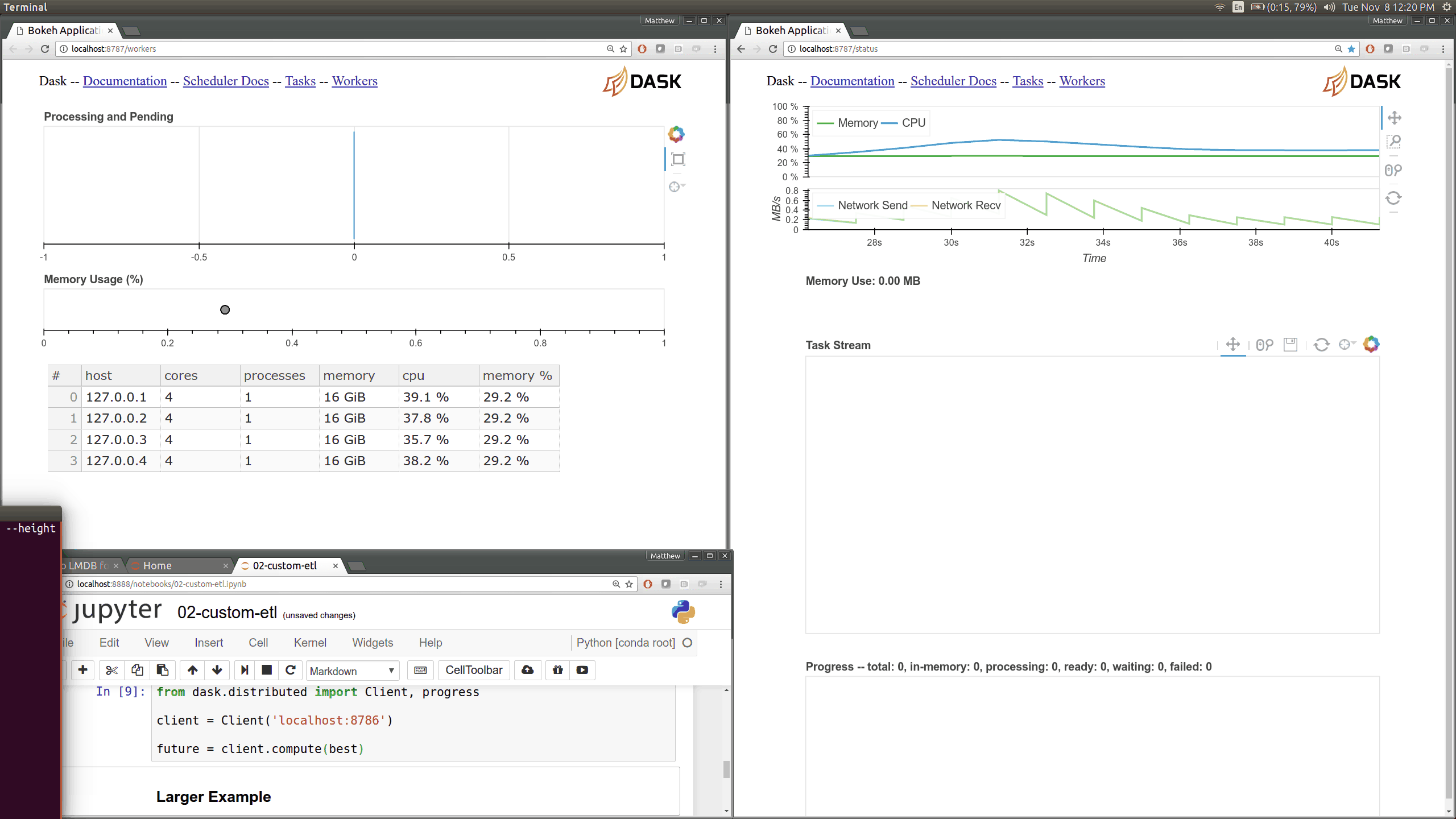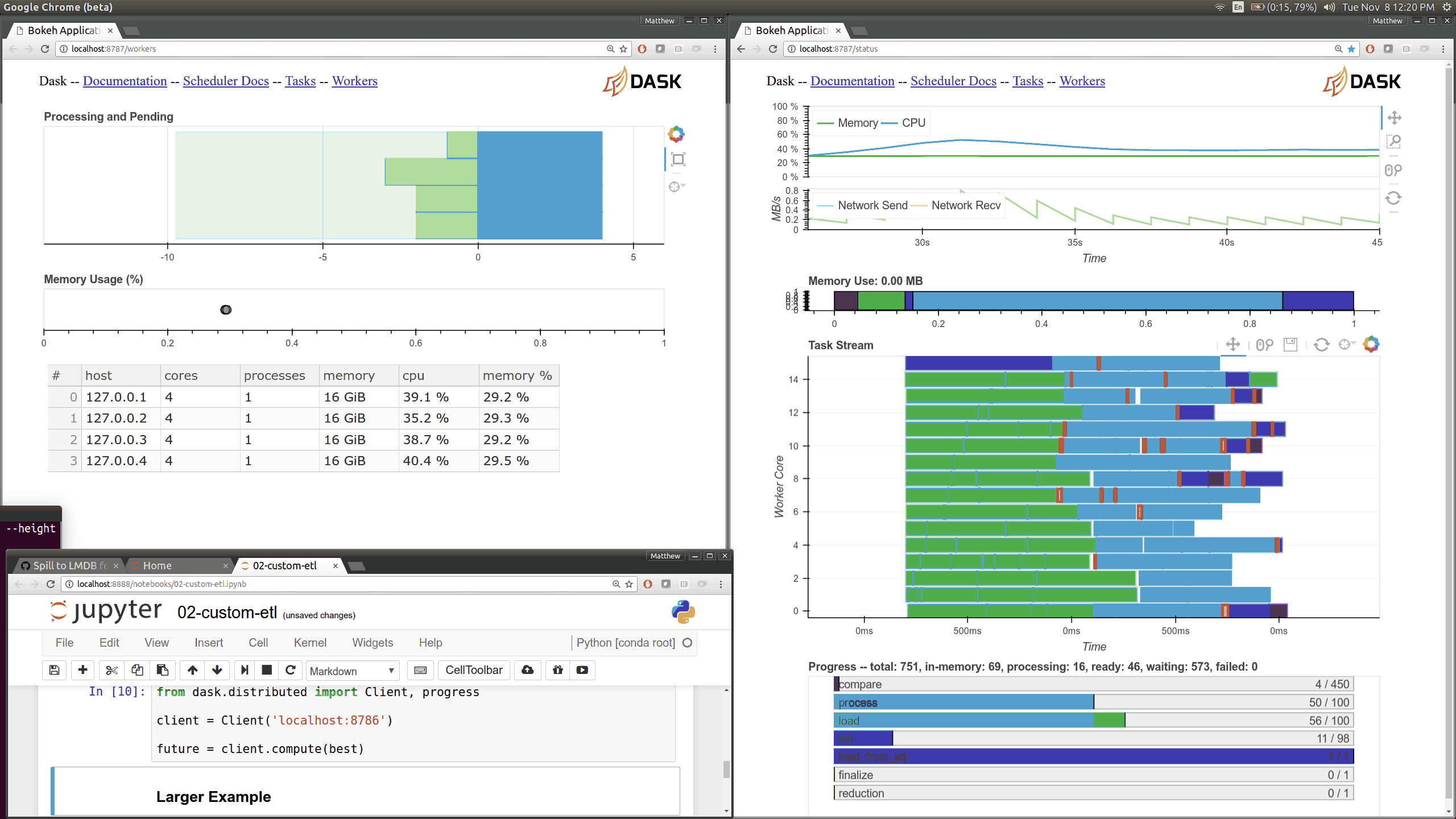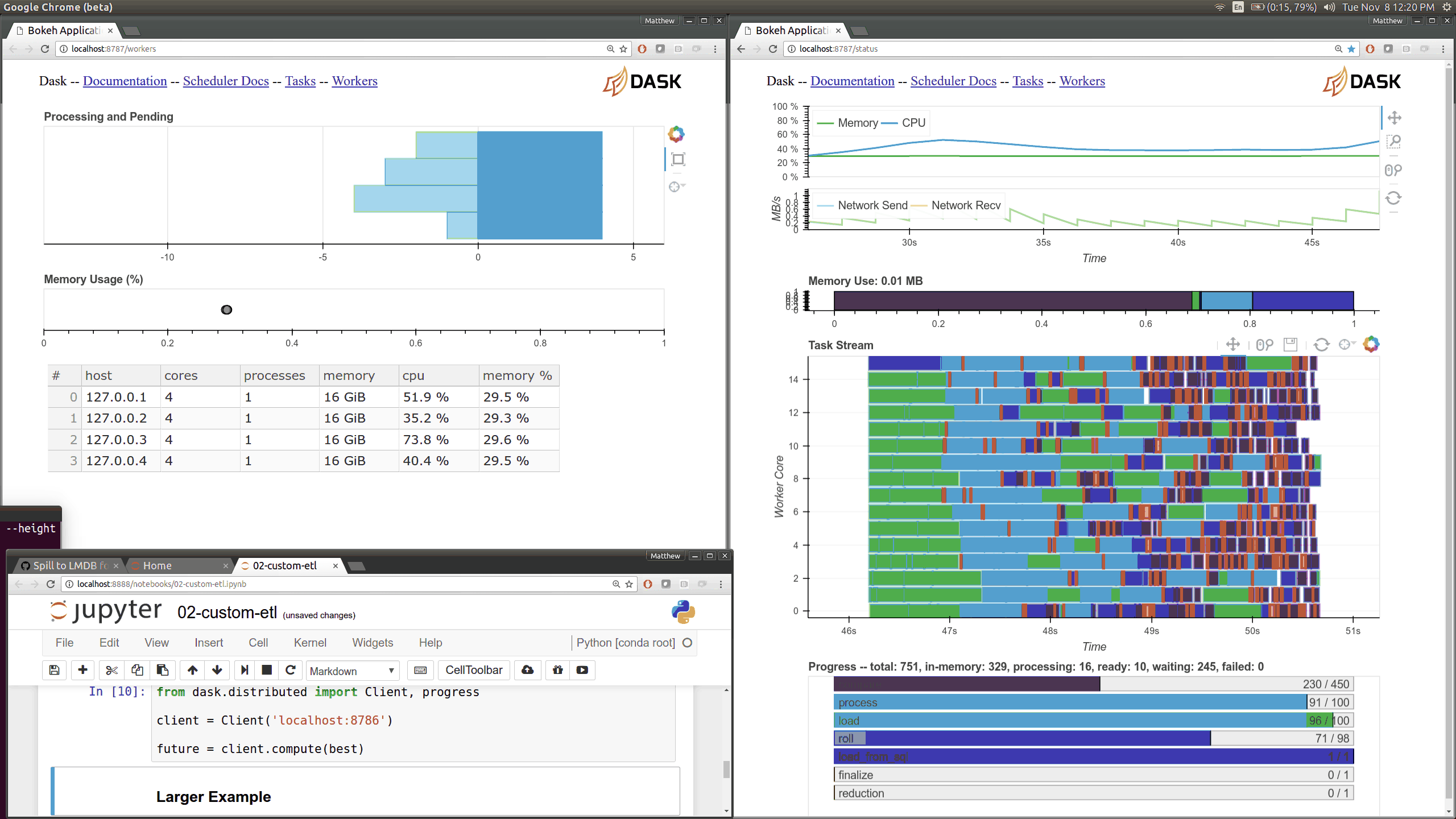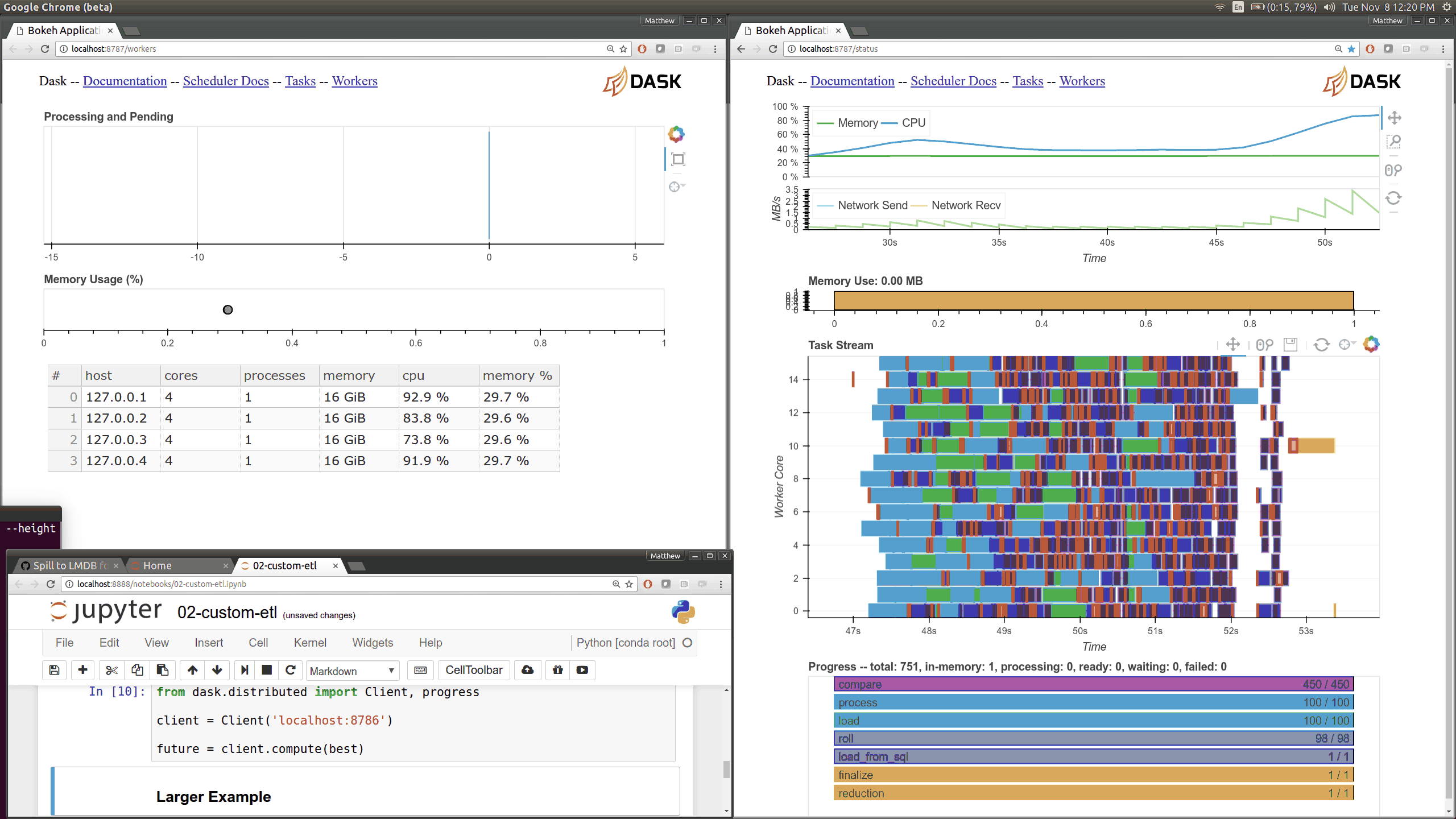
By default, when no custom Client is specified, Arboretum creates a LocalCluster instance with the diagnostics dashboard disabled:
...
local_cluster = LocalCluster(diagnostics_port=None)
client = Client(local_cluster)
...
You can easily create a custom LocalCluster, with the dashboard enabled, and pass a custom Client connected to that cluster to the GRN inference algorithm:
local_cluster = LocalCluster() # diagnostics dashboard is enabled
custom_client = Client(local_cluster)
...
network = grnboost2(expression_data=ex_matrix,
tf_names=tf_names,
client=custom_client) # specify the custom client
By default, the dashboard is available on port 8787.
For more information, consult:
- Dask web interface documentation
- Running with a custom Dask Client
Q: My gene expression matrix is transposed, what now?¶
The Python scikit-learn library expects data in a format where rows represent observations and columns represent features (in our case: genes), for example, see the GradientBoostingRegressor API.
However, in some fields (like single-cell genomics), the default is inversed: the rows represent genes and the columns represent the observations.
In order to maintain an API that is as lean is possible, Arboretum adopts the scikit-learn convention (rows=observations, columns=features). This means that the user is responsible for providing the data in the right shape.
Fortunately, the Pandas and Numpy libraries feature all the necessary functions to preprocess your data.
\ Sort by:\ best rated\ newest\ oldest\
\\
Add a comment\ (markup):
\``code``, \ code blocks:::and an indented block after blank line